Fixing PCR errors in sequencing experiments
Published in Bioengineering & Biotechnology
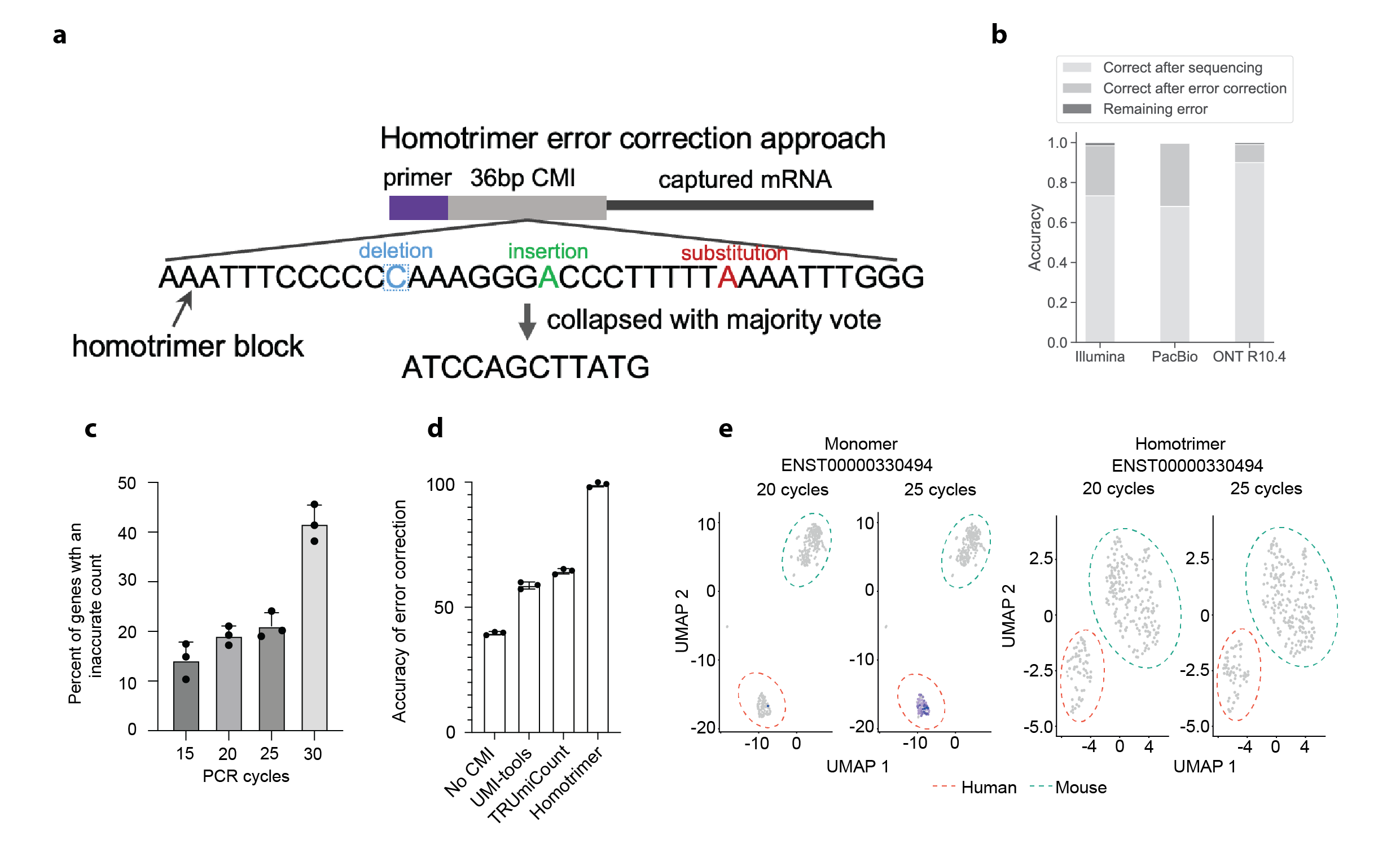
Accurate quantification of RNA molecules is essential for elucidating cellular heterogeneity in single-cell sequencing data. The integration of unique molecular identifiers (UMIs) into transcripts during library preparation has become the method of choice for counting RNA molecules. This approach is particularly effective for demultiplexing cell barcoded-reads in downstream analysis. The efficacy of this method can be further enhanced by employing computational algorithms. However, the effectiveness of both experimental and computational strategies is significantly challenged when the number of PCR artefacts, particularly those with error-prone UMI regions, increases with the number of PCR cycles. This limitation underscores the need for improved experimental methods to accurately correct for PCR-amplified artefacts.
- Findings
Inspiration from information theory
Drawing inspiration from cryptographic techniques, where messages are sent multiple times and validated through a majority vote to ensure accuracy, we developed an innovative approach for UMI error correction. We designed a novel UMI structure composed of homotrimer blocks, termed as homotrimer UMIs (Figure 1A). This design allows us to collapse homotrimer blocks by using a “majority voting” strategy, achieving error correction within building blocks of UMIs prior to further grouping UMIs wherever possible. This method aligns with principles in information theory, particularly in evaluating the entropy of a character string. Here, it is applied to assess how information in each triplet of nucleotides is altered throughout PCR amplification and sequencing. A triplet that remains consistent yields the lowest entropy, whereas variability within a triplet's nucleotides results in the highest entropy.
Determination of the major source of UMI errors
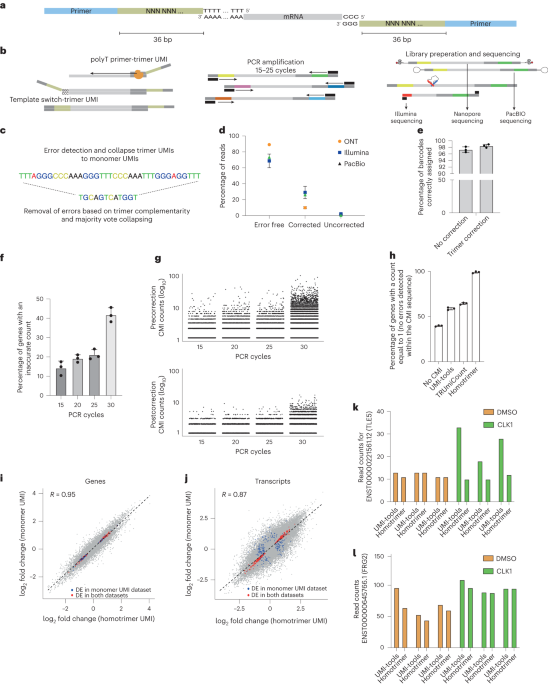
To address the challenges in quantifying the impact of unique molecular identifiers (UMIs) on molecular counting, due to their random synthesis and unknown composition, we experimented with a known 36-nucleotide homotrimer common molecular identifier (CMI). This CMI was integrated into the 3’ end of human and mouse cDNA, allowing its reduction to a 12-nucleotide UMI compatible with V3 chemistry. This approach ensures accurate transcript counting, contingent on error-free CMIs, and aims to provide a more precise evaluation of UMI efficiency and effectiveness in molecular counting. Using the CMI we were able to show that UMI errors are the main source of UMI errors in both bulk and single-cell sequencing.
Near-absolute counting of RNA molecules
By incorporating homotrimer UMIs prior to PCR amplification, we demonstrated the ability to eliminate errors, thereby enabling nearly absolute counting of RNA molecules. Our analysis, conducted across various sequencing platforms – Illumina, PacBio, and the latest Oxford Nanopore Technologies (ONT) – revealed that the proportions of correctly identified common molecular identifiers (CMIs) were 73.36%, 68.08%, and 89.95%, respectively. When applying our majority voting strategy, these accuracies significantly improved to 98.45%, 99.64%, and 99.03%, respectively, leading to a minimal error rate in sequenced reads (as shown in Figure 1B). At the single-cell level, this methodology similarly corrected CMI-tagged PCR duplicates with an accuracy range of 96-100%. Overall, the homotrimer error correction strategy effectively achieves near-absolute RNA molecule counting.
The application of the homotrimer approach in biological research – a case study
To demonstrate the biological significance of adopting the homotrimer approach, we conducted a case study focusing on the identification of biological pathways in differentially expressed genes. These genes were detected using both homotrimer and monomer UMIs. Our findings indicated that error correction applied to homotrimer UMIs eliminated false positive differentially regulated genes from the output of downstream analyses in both bulk and single-cell sequencing experiments, thereby ensuring more accurate and reliable results.
Conclusion
Our homotrimer UMIs have been developed to discriminate true transcripts from PCR duplicates and verified both experimentally and computationally useful to improve RNA molecule counting accuracy across multiple sequencing platforms at both bulk and single-cell levels. We have identified PCR-induced errors as the main error source in both RNA-seq and single-cell RNA-seq and our proposed homotrimer error correction approach achieves the leading advance in improving UMI quantification accuracy over the current state-of-the-art methods.
You can access our Nature Methods paper here.

Homotrimer error correction. A. Illustration of the homotrimer UMI and the “majority voting” error correction. B. Accuracy of RNA molecule counting using CMIs across Illumina, PacBio and ONT sequencing platforms.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in