From Mutation to Peptide: Resolving Proteome Complexity with moPepGen
Published in Bioengineering & Biotechnology, Cancer, and Protocols & Methods
Explore the Research
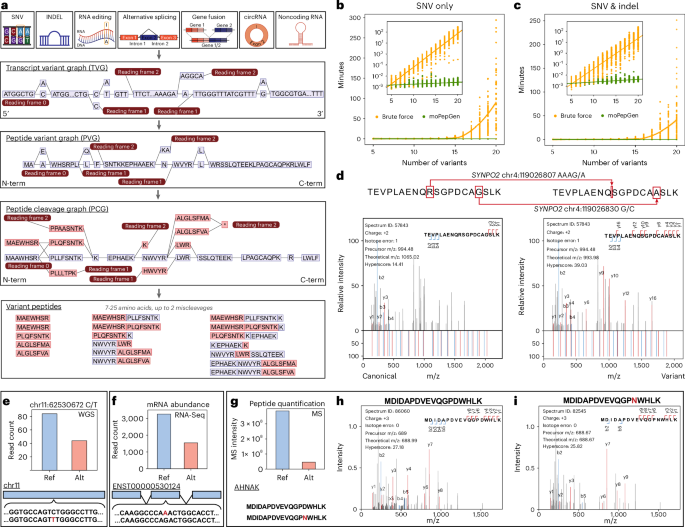
Identification of non-canonical peptides with moPepGen - Nature Biotechnology
moPepGen enables the detection of peptides across species, proteases and technologies.
Proteogenomics is changing how we study gene expression. It connects information from DNA and RNA to the proteins actually produced in cells. By integrating genomics, transcriptomics, and proteomics, researchers can now investigate gene expression at two distinct levels: the quantity of proteins produced and the sequence diversity of those proteins, shaped by genomic and transcriptomic variation. This latter dimension is especially important in cancer, where gene expression is distorted by mutations, alternative splicing, fusion events, and a wide range of RNA-level changes. Proteogenomics has already helped identify new cancer subtypes, improve patient stratification, and discover therapeutic targets. But even with its successes, much of the human proteome remains hidden.
The primary reason lies in the reliance of mass-spectrometry-based proteomics on database search strategies. Most workflows query spectral data against reference protein databases that exclude sample-specific genomic and transcriptomic variants. Consequently, peptides resulting from somatic mutations, aberrant splicing, non-canonical open reading frames, and other transcriptomic alterations are often missed.
A natural solution would be to incorporate sample-specific variants into the search space. However, this presents a computational and biological challenge: exhaustively enumerating all possible combinations of variants within a gene quickly becomes infeasible due to the exponential explosion of potential sequences. Most existing tools either restrict their focus to single variant types or assume that multiple variants do not co-occur on the same transcript. This assumption is biologically unrealistic. In practice, transcripts often carry combinations of SNVs, indels, splicing variants, and other transcriptomic variations. Ignoring these combinations severely limits our ability to detect non-canonical peptides and constrains our understanding of the proteome.

From variant to peptide: moPepGen enables comprehensive and scalable proteogenomic analysis.
To address this gap, we developed moPepGen, a graph-based algorithm that constructs comprehensive peptide databases reflecting the full spectrum of variant combinations observed in an individual sample. moPepGen takes a transcript-centric approach: all detected genomic and transcriptomic variants are integrated into a single directed acyclic graph per transcript. Translation and in silico digestion are then performed directly on the graph.
This strategy offers significant computational and biological advantages. The graph representation reduces redundancy, preserves the context of co-occurring variants, and supports linear runtime scalability to the number of variants. moPepGen systematically models a wide range of alterations, including substitutions, indels, alternative splicing, gene fusions, RNA editing and circular RNAs. Benchmarking demonstrates that moPepGen predicts approximately four times more non-canonical peptides and identifies about twice as many of them compared to prior methods.
We demonstrated moPepGen across diverse datasets and biological systems. In human cancer cell lines and primary tumors, it enabled the detection of thousands of non-canonical peptides that were not captured by conventional methods. In the mouse proteome, it successfully recovered unannotated novel translation products. moPepGen is compatible with both data-dependent (DDA) and data-independent (DIA) acquisition and can be seamlessly integrated into multi-omics pipelines combining genomic, transcriptomic, and proteomic data.
This tool is designed for practical use. It has native support for output files directly from upstream variant calling algorithms and generates non-canonical databases rapidly. Its modular architecture enables flexibility in the types of variants and the algorithms used to detect them. moPepGen is fully open-source, thoroughly documented, and backed by an extensive suite of unit tests and fuzz tests that ensure the accuracy and robustness of variant peptide calling.
moPepGen was developed to bridge a critical gap in proteogenomics. Rather than simplifying biological complexity to accommodate computational constraints, we built an algorithm that accommodates biological reality. We anticipate that moPepGen will empower researchers to explore the hidden layers of the proteome with greater resolution and biological fidelity.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in