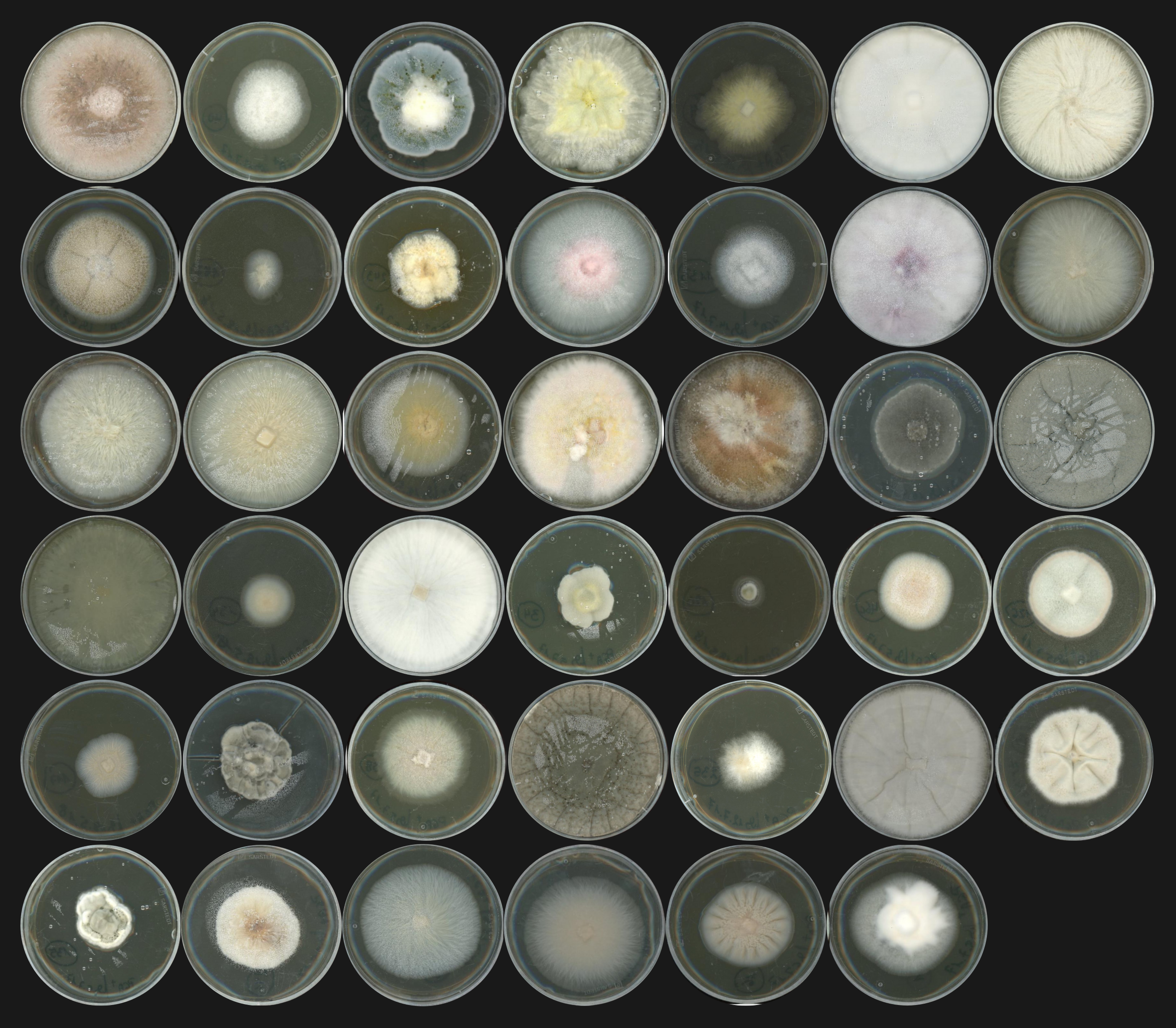

Complex microbial consortia — including bacteria and fungi — inhabit plant roots and influence the health and development of their host. Although the mechanisms underlying the assembly of these communities remain poorly understood, the root microbiome has been better characterized over the last decade. Surveys of its composition across different plant species and environments were made possible by an increased affordability of microbial profiling, with the ribosomal DNA amplicon sequencing method. Additionally, the development of experimental approaches with synthetic microbial communities (SynComs) allowed to disentangle the effects and functions of the root microbiota.
Between 2013 and 2017, Dr. Stéphane Hacquard and his group at the Max Planck Institute For Plant Breeding Research (MPIPZ - Cologne, Germany) undertook a continental-scale sampling of Arabidopsis thaliana plants. By profiling the root microbiota in 17 natural populations of the model species between Spain and Sweden, the researchers identified climate and soil properties as the main drivers of community differentiation for filamentous eukaryotes and prokaryotes, respectively (Thiergart et al., 2020). A culture collection of root-associated bacterial and fungal strains isolated along the same European transect was also assembled. With this resource in hand, the group could design SynComs and perform plant recolonization experiments in gnotobiotic culture systems. This approach notably highlighted that the fungal component of the microbiota — the mycobiome — has a high pathogenic potential, and can severely affect plant health when protective bacterial commensals and plant immune outputs fail to restrain fungal proliferation in plant roots (Durán et al., 2018 ; Wolinska et al., 2021).
In April 2015, the microbiologist and plant physiologist Dr. Francis Martin — research group leader at the French National Institute for Agriculture, Food and Environment (INRAE - Nancy, France) — was invited by S. Hacquard to give a seminar at MPIPZ. Investigating mycorrhizae from the genomic perspective, F. Martin has become over the years a world-renowned expert on symbiotic associations between plant roots and fungi. S. Hacquard took the opportunity of his visit to show him the culture collection of A. thaliana root-associated fungi he was assembling. This plant species, as other Brassicaceae, is unable to associate with beneficial mycorrhizal fungi to acquire nutrients, since it does not harbor the genetic network needed to establish a so-called mycorrhiza. The root-associated fungi of non-mycorrhizal plants and the genetic determinants allowing them to colonize their host remained to be characterized. F. Martin informed S. Hacquard that the sequencing of these fungi could be integrated in his proposal “1KFG: Deep Sequencing of Ecologically - relevant Dikarya”, that he was planning to submit the following day to the U.S. Department of Energy Joint Genome Institute (JGI - Walnut Creek, California, USA) as part of the Community Science Program (CSP) annual call. After a long day spent writing a section in the proposal, the genome sequencing of 41 representative A. thaliana root mycobiota members could be integrated just on time before submission. As this project was accepted a few months later, a collaborative study aiming at identifying genomic signatures underlying the association of these fungi to A. thaliana roots started between the groups of S. Hacquard, F. Martin and Dr Igor Grigoriev (JGI). It is now published in Nature Communications (Mesny et al., 2021).

When I — Fantin Mesny — started my PhD in S. Hacquard’s group in December 2018, I had the chance that the last of the 41 genomes sequenced at the JGI were being assembled and annotated. The strains had been carefully selected by my new supervisor and post-doctoral researcher Thorsten Thiergart, who used their microbial profiling data (Thiergart et al., 2020) to choose, in a collection of about 600 isolates, 41 strains corresponding to the most representative fungal taxa, often detected in the roots of wild European A. thaliana populations. Most of the fungal biodiversity in the root endosphere could not be easily isolated and cultured in vitro, but the taxa of the 41 selected strains covered 35% of the cumulative fungal relative abundance across sites identified by amplicon sequencing. Focusing on these isolates, I could use comparative genomics and plant recolonization experiments to decipher the evolution and function of the A. thaliana root mycobiota.
In the aim of better characterizing the association between mycobiota members to plant roots, I compared the newly sequenced fungi to 79 closely related strains with published genomes. I classified these reference fungi into lifestyle categories — mycorrhizal fungi, saprobes, endophytes and pathogens —, by referring to their isolation source, the FunGuild database and the expertise of F. Martin. Post-doctoral researcher Dr. Shingo Miyauchi and myself compared the resulting lifestyle categories, focusing on their composition in genes known to be involved in fungal association to hosts and environment: carbohydrate-active enzymes, effector-like small secreted proteins, proteases and lipases. We identified strong similarities between our mycobiota members and well-characterized endophytes. An analysis by confocal microscopy of plants recolonized with six of our isolates confirmed that these fungi can endophytically colonize the cortex of A. thaliana roots.
")
When the coronavirus pandemics hit Germany and the first lock-down happened, I had to pause my experiments, and focused on the parts of the project I could work on from home. At that time, we aimed at identifying gene families that could be important for endophytism, in both our newly sequenced genomes and reference endophytes. Difficulties I was facing were the size of the data set (41,612 gene families) making multiple-testing difficult, and its phylogenetic signal (3 phyla, 7 classes). I decided to dedicate a few of weeks of teleworking to read and learn about machine learning methods. When doing so, I realized how I could extract from an orthology prediction the minimal set of gene families that best segregates categories of organisms — in my case endophytes and mycobiota members from other fungal lifestyles. With this approach, I identified a set of 83 gene families that constitute putative genetic determinants for endophytism, including plant cell wall-degrading enzymes that we later found over-expressed in planta by six phylogenetically-distant mycobiota members (lytic polysaccharide monooxygenase AA9 and xylanase GH10). These weeks of exploration also inspired me to use another type of machine learning models to predict the ancestral lifestyles of our mycobiota members. Interestingly, ancestors were not endophytic, and most resembled plant pathogens.
To better understand this result and decipher the previously described pathogenic potential of the A. thaliana mycobiome, plants grown in a closed system in absence of any micro-organism were recolonized with our 41 individual mycobiota isolates. With this experiment, I identified a wide diversity of fungal effects on plant growth, ranging from highly detrimental to beneficial. These were shown to be correlated to fungal colonization ability — both to the relative fungal abundance in natural populations and to the fungal burden measured in roots from our gnotobiotic experiments. Best colonizers displayed the highest pathogenic potential.
In the hope of identifying candidate genes explaining the strong colonization ability resulting in detrimental effects on plant health, I associated fungal effects to genome compositions with different approaches, including a machine learning model. Two independent methods identified a gene family that could explain detrimental effects and root colonization abilities. This family (pectate lyase PL1_7) encodes enzymes that degrade pectin, an essential constituent of plant cell walls, which is especially abundant in the roots of dicotyledonous plants like A. thaliana. To validate its involvement in root colonization and pathogenicity, we decided to collaborate with Dr Lea Atanasova (University of Natural Resources and Life Sciences - Vienna, Austria) and Dr Magnus Karlsson (University of Agricultural Sciences - Uppsala, Sweden). For one of their previous studies, they had introduced a gene from family PL1_7 in the genome of a fungal species which naturally does not have any, resulting in an increased pectin degradation efficiency of the mutant strain. Plant recolonization experiments performed with their wild-type and mutant strains highlighted that the PL1_7 gene conferred an increased root colonization ability to the fungus, simultaneous to a significant decrease of the performance of inoculated plants.
Therefore, mycobiota members — and more generally, endophytes — colonize A. thaliana roots by degrading plant cell walls, using notably enzymes from family PL1_7. In the light of previous studies (Durán et al., 2018 ; Wolinska et al., 2021), we can expect both bacterial root commensals and the plant immune system to contribute in protecting roots from a detrimental over-colonization of fungi. A question that remains to be addressed is whether a different set of carbohydrate-active enzymes is required to colonize endophytically other hosts with different cell wall compositions.
I feel very lucky I got to work as a PhD student on this project. I am thankful to all scientists involved, and especially to my supervisor S. Hacquard for his trust and letting me explore ideas and methods that ended up constituting a significant part of this study. Collaborations made this project possible, and allowed its successful course from ecological data to the validation of a candidate gene. This project could not have been possible without the funding from the DFG Priority Programme 2125 “Deconstruction and Reconstruction of the Plant Microbiota –DECRyPT” and from the European Research Council Starting Grant MICRORULES. We hope our results, together with our new set of genomes — now publicly available — will open the way for new studies on endophytism, and contribute to a better understanding of the fine line between endophytism and parasitism in the fungal root microbiome.
References
Durán, P. et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973-983.e14 (2018).
https://doi.org/10.1016/j.cell.2018.10.020
Mesny, F. et al. Genetic determinants of endophytism in the Arabidopsis root mycobiome. Nat. Commun. (2021).
https://doi.org/10.1038/s41467-021-27479-y
Thiergart, T. et al. Root microbiota assembly and adaptive differentiation among European Arabidopsis populations. Nat. Ecol. Evol. 4, 122–131 (2020).
https://doi.org/10.1038/s41559-019-1063-3
Wolinska, K. W. et al. Tryptophan metabolism and bacterial commensals prevent fungal dysbiosis in Arabidopsis roots. Proc. Natl. Acad. Sci. U. S. A. 118, 2111521118 (2021).
https://doi.org/10.1073/pnas.2111521118
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in