Genomic rearrangements involving non-coding regions alter gene regulation in cancer
Published in Cancer
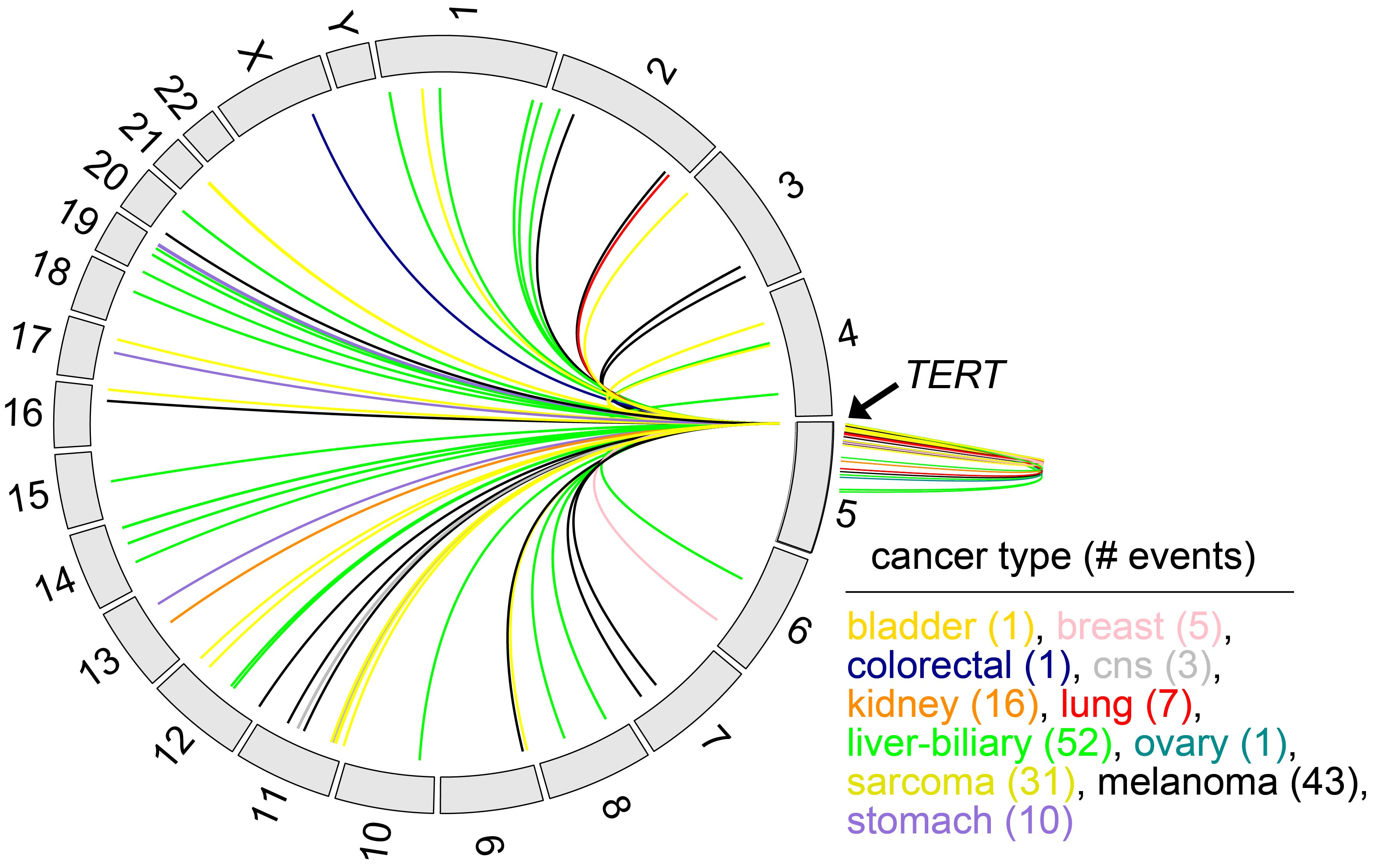

In late 2013, as co-chair of the kidney chromophobe project sponsored by The Cancer Genome Atlas (TCGA) consortium, I got to take part in a serendipitous discovery. Our team was collectively analyzing 50 kidney chromophobe cancer genomes by whole genome sequencing. This was at a time when much of the focus of cancer genomics projects was on mutations occurring within the exome, which represents just ~1% of the entire genome. By gene expression analysis, we found that the TERT gene, which encodes a catayltic subunit of the enzyme telomerase and is thereby able to confer unlimited proliferation potential to cancer cells, was expressed at very high levels for a subset of our cancer cases. However, we found no alterations within the gene itself that would explain this. Looking at alterations outside of TERT by whole genome sequencing analysis, specifically within the region 20kb upstream of the gene, we found evidence of structural variant (SV) breakpoints involving the same cases with high TERT expression. Soon after our TCGA study, subsequent studies could observe SV-associated up-regulation of TERT for other cancer types.
We now know of multiple mechanisms by which genomic rearrangements may lead to altered cis-regulation, depending on the specifics of how the rearrangements might change the regulatory landscape for a given gene. Regulatory element disruption, enhancer hijacking, disruption of topologically-associated domains (TADs), and altered DNA methylation are all potential mechanisms (illustrated below). SVs of all classes (translocations, tandem duplications, deletions, etc.) may be involved in altering gene regulation.

As the kidney chromophobe study was wrapping up, I was invited to join the PCAWG consortium, which aimed to aggregate whole genome sequencing data on over 2500 human cancers from TCGA and the International Cancer Genomics Consortium (ICGC). Of these cases, 1220 also had corresponding gene expression data. This presented an opportunity for us to examine SV-mediated gene deregulation in a very large cohort of multiple cancer types. We suspected that other genes in addition to TERT could be impacted by this phenomenon. For the PCAWG study, we developed an analytical approach to systematically examine all genes for altered expression in the presence of nearby somatic SV breakpoints. We found that lots of genes are impacted in this way, independently of any associated copy number alterations. Cancer-associated genes of interest included TERT, MDM2, CDK4, ERBB2, CD274, PDCD1LG2, and IGF2. TERT-associated breakpoints involved ~3% of cancer cases, most frequently in liver biliary, melanoma, sarcoma, stomach, and kidney cancers. Similar to TERT, other oncogenes may be affected in a small percentage of cases, but the cumulative effect of the many genes affected greatly extends the total numbers of patients that would be involved.
Our study benefited from being part of the PCAWG consortium. Participation in big science team efforts has both advantages and disadvantages for the individual investigator. With PCAWG, we had access to high quality genomic and transcriptomic datasets, which would have taken my group an incredible amount of time to generate on our own. Before submission, the PCAWG Steering Committee members reviewed our manuscript and provided helpful comments, which improved it considerably. On the other hand, the timeline from manuscript writing to publication of our paper was quite long (the manuscript was first drafted in early 2017, and the paper was published in 2020), in large part because of the need for all the PCAWG papers to be coordinated with each other. However, in the meantime, through other recent papers, we have been able to examine this phenomenon in other patient cohorts, as well as demonstrate a similar phenomenon of altered DNA methylation in relation to nearby somatic SV breakpoints.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in