Great or small, Legionella can secrete it all
Published in Microbiology

Gram-negative bacteria utilize a wide range of trans-membrane machineries, called secretion systems, to translocate proteins into host eukaryotic cells. These so called “bacterial effector proteins” interfere with host cell functions. Elucidating the mechanism of action of secretion systems will therefore provide a basis for novel antibiotics design strategies to combat bacterial infectious diseases.
Our model system is the gram-negative bacterium Legionella pneumophila, the causative agent of Legionnaire disease. This pathogen translocates over 300 effector proteins (most pathogenic bacteria settle for less than a dozen) into alveolar macrophages, by utilizing the Dot/Icm type IV secretion system. L. pneumophila employs an effector recruitment platform, an inner membrane complex called the Type IV Coupling Complex (T4CC), that recruits effectors and translocates them through the secretion channel. How the T4CC orchestrates the recruitment and transfer of so many different effectors is still unclear.
We were fascinated by this functionally essential complex, and in order to elucidate its mechanism of action, we sought to solve its structure. Our first approach, to purify the complex recombinantly in Escherichia coli, failed despite extensive attempts. A solution was found when we introduced an affinity tag to the main component of the T4CC, the AAA+ ATPase DotL, in the L. pneumophila genome. This enables the purification of a stable complex from Legionella cells. From there, the purified machine revealed more surprising findings as study progressed. First, a complex of three proteins was expected but the purified complex was composed of eight components. Secondly, two components, never reported before, were discovered bound to the complex, and were named DotZ and DotY. The third surprising outcome was the T4CC structure and the way it assembles (Fig. 1). Indeed, DotL has a very long C-terminal tail onto which all other components assemble like pearls on a string. At the end of the tail, the effector-recruiting module made of IcmS and IcmW (IcmSW) dangles in various conformations. Because AAA+ ATPases function as channel-forming hexamers, we proposed a hexameric model of the entire T4CC based on a homologue of DotL. Interestingly, the various conformations of the IcmSW module delineate a trajectory that would deliver a bound effector to the ATPase channel (Fig.1).
In conclusion, when starting this project, we never imagined it will take us so far (for one of the first co-authors, it also meant flying across the pond several times to carry out some of the experiments in Yale). But the T4CC turned out to be a lot more fascinating than we anticipated. It suggested different binding regions for different classes of effector, provided us with information on the dynamics of the system, and overall enhanced greatly our understanding of effector recruitment platforms in secretion systems.
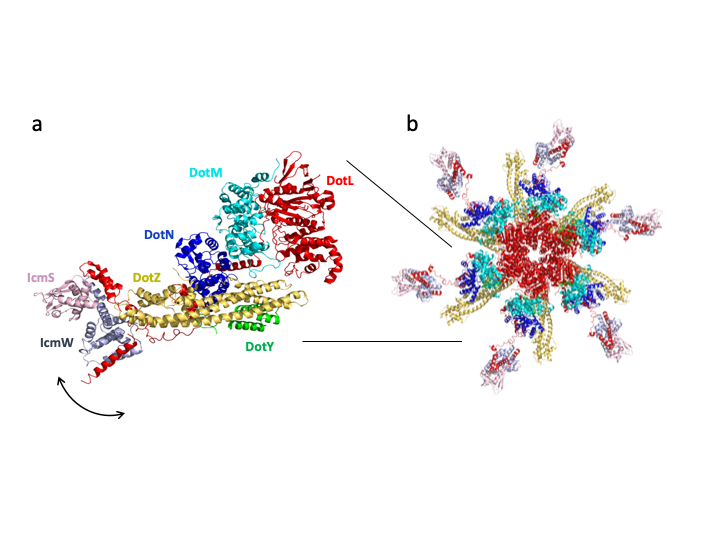
Figure 1. The structure of the T4CC. a. Ribbon diagram of the T4CC . All proteins are indicated in a different colour and labelled. The arrow indicates the positional flexibility of the IcmSW module relative to the heteropentameric DotLMNYZ core. b. Hexameric model of the T4CC. The lines indicate the position of the DotLMNYZ-IcmSW complex shown at left.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in