Higher order genetic interactions switch cancer genes from two-hit to one-hit drivers
Published in Cancer
The genes that cause cancer when they are mutated – called ‘cancer driver genes’ – can be classified as oncogenes or tumor suppressor genes depending upon whether their activation or inactivation contributes to cancer. In our genome, we have two copies of every gene – one from each parent. Classically, a single mutation in an oncogene can be sufficient to increase tumor fitness, whereas inactivation of both copies of a tumor suppressor gene is often required, a concept originally called the ‘two-hit hypothesis’ 1,2. However, we live in the ‘genomics’ era, where huge amounts of sequencing data from cancer patients have accumulated. With so much ‘big data’ available, many exceptional cases beyond the classical models are coming to light 3,4. For example, some oncogenes are dosage-sensitive, with amplification of a mutated copy further increasing tumor fitness 5-7 and some tumor suppressor genes are haploinsufficient, with inactivation of a single allele promoting cancer 3,8-10.
In our recent paper, we asked two simple questions: (1) how diverse are the interactions between somatic mutations and copy-number alterations within a single gene in different contexts, specifically in different types of cancer and in different genetic backgrounds, (2) why do the interactions within a gene change in different tumors and contexts. These questions started several years ago when I was conducting postdoctoral research in Ben Lehner's lab at the Centre for Genomic Regulation (CRG) in Barcelona and Fran Supek had just started his own lab at Institute for Research in Biomedicine (IRB) also in Barcelona, Spain. I had just finished work quantifying the interactions between genomic alterations in different cancer genes – how they cooperate or act redundantly and how this changes across tissues11 - and how interactions between inherited genetic variants and changes in copy number in tumors can be used to identify new cancer predisposition genes 12. We wanted to combine these two approaches of quantifying how genetic changes interact between genes and in the same gene: does whether a change in copy number of a mutated cancer gene contributes to cancer depend upon the other cancer genes that are mutated in a tumor? This would require quantifying ‘higher order’ interactions between mutations. We would have to not only quantify whether what happens with two mutations is different from the sum of their individual effects but also whether what happens with three genomic alterations is more than their individual effects and pairwise interactions. So, this is a part of our long journey together to explore genetic interactions – how mutations interact - to improve our understanding of tumorigenesis.

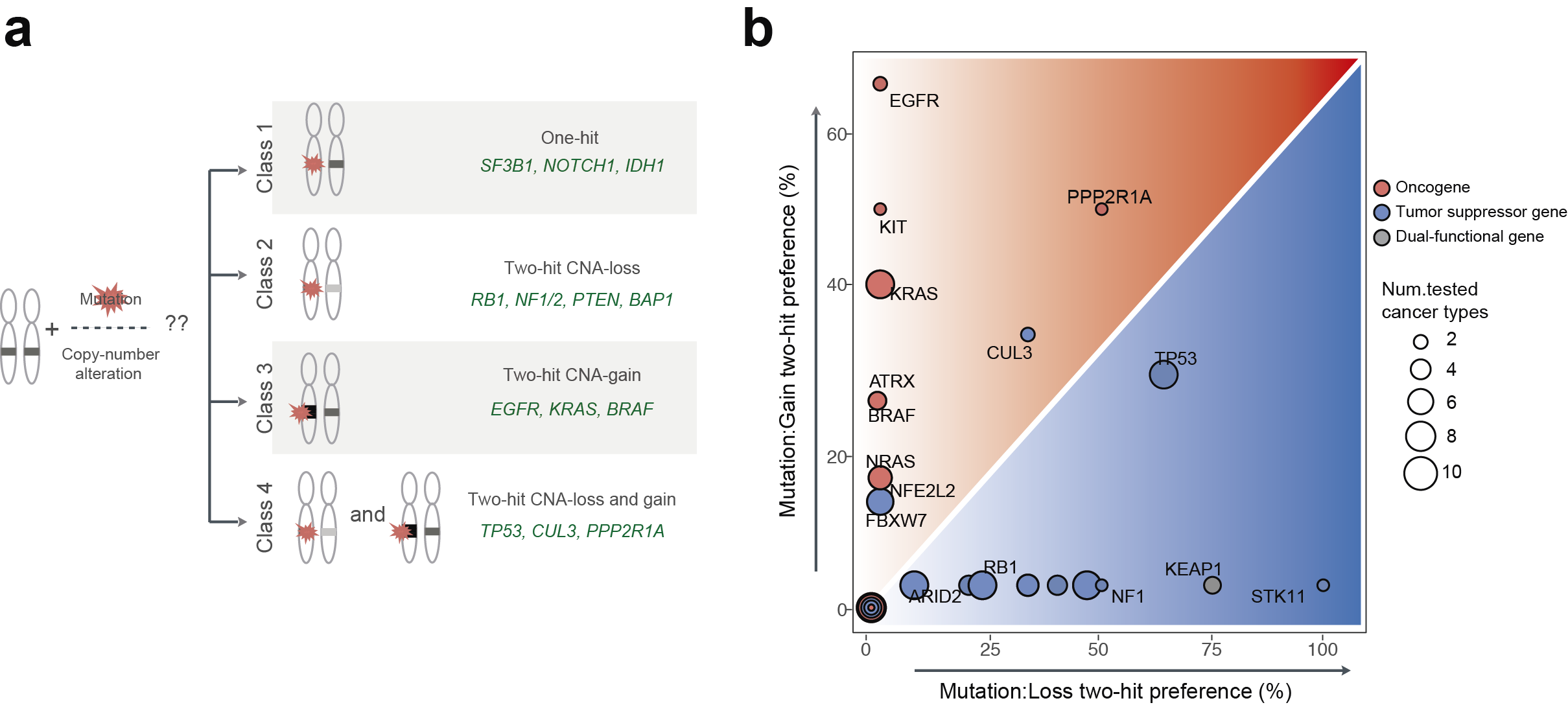
Fig1. Interactions between somatic mutations and copy-number alterations in human tumors. (a) Four classes of cancer genes are based on their interaction preferences between mutations and copy-number changes. Class 1 (one-hit driver) which is no significant co-occurrence between mutation and CNAs in cancer types in which they are significantly mutated. The other classes show strong interactions between mutations and copy-number loss (class2; two-hit loss) or copy-number gain (class3; two-hit gain). Class 4 (two-hit loss and gain) has interactions between mutations and both CNAs-loss and gain depending on cancer types. Represented cancer genes in each class are presented with green. (b) Frequency of interaction between mutation and copy-number loss (x-axis) and copy-number gain (y-axis) in all cancer types in which each gene is mutated in >2% of samples. Color indicates the type of cancer genes (blue: tumor suppressor gene, red: oncogene, grey: dual-functional gene).
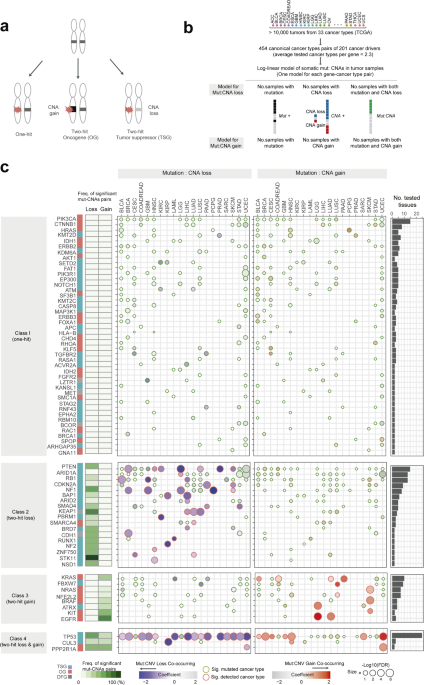
To answer these questions, we analyzed large-scale cancer genomics data from The Cancer Genome Atlas (TCGA) project. We quantified the interactions between mutations and copy-number changes within a gene across more than 30 cancer types. We began by showing that interactions between mutations and copy-number alterations of 201 cancer genes are diverse across cancer types. Based on the data we suggest four classes of cancer genes. Class 1 genes are ‘one-hit drivers’: cancer genes where a single mutation seems enough to contribute to cancer and there is no or little benefit of amplifying the mutated gene or deleting the other copy. Both tumor suppressor genes and oncogenes can be class 1 drivers. The other classes show strong interactions between mutations and copy number changes. The interactions are with copy number loss (class 2; 'two-hit loss') or copy number gain (class 3; 'two-hit gain'). Each class is mainly composed of tumor suppressor genes (class 2) or oncogenes (class 3). Class 4 (two-hit loss and gain), curiously, consists of genes that have interactions between mutations and both copy number loss and gain, depending on the tissue; this class includes the master tumor suppressor gene TP53.
Although the class 4 cancer genes present extreme examples of the interactions between mutations and copy-number alterations varying across cancer types, the data suggest that this might also happen for some other drivers in the other classes, although the statistical trends are more subtle. For example, whereas mutations in the classic tumor suppressor gene NF1 interact with copy-number alteration loss in most cancer types in which the gene is mutated, the driver mutations in BRAF specifically interact with copy-number alteration gain in only one of four cancer types (skin cancer) in which the gene is highly mutated. We further explored changes in these interactions across cancer types by testing whether the strength of interaction between mutations and copy-number alterations differs between cancer types. We found that many of the interactions (~40%) present clear differences in strength between cancer types, meaning that the tissue dictates how cancer genes interact.
The next question was why the interactions between mutations and copy-number alterations change in different cancer types? One cause could be higher-order genetic interactions. We could imagine the simplest example, where the genetic interaction between two mutations can be changed depending upon the presence of a third mutation (a so-called ‘third-order interaction’). These higher-order interactions have been shown to be important in model organisms and in individual proteins and RNAs, and we hypothesized that conceptually similar higher-order interactions might be occurring in human tumors. We devised a method to test for these 3-way interactions in cancer genomes and indeed found clear examples where the interactions between a mutation and copy-number alteration in a gene (the first gene with two genomic alterations) can be altered by the mutations in a second gene. Because many tumor patients carry more than two known driver mutations (and likely additional yet-undiscovered ones), there is plenty of opportunity for higher-order interactions between different cancer genes.
With a total of 1,511 pairwise interactions tested for 40 genes, we identified 17 third-order interactions (FDR 10%) either positive (the presence of a mutation in a second gene increases the strength of the interaction of the mutations in the first gene) or negative (the presence of a mutation in a second gene decrease the strange of the interaction). Interestingly, most of the third-order interactions involve two genes from the same signaling pathway, including the PI3K pathway (breast and uterine cancer), RTK/RAS pathway (skin cancer), NRF2 pathway (lung squamous cell carcinoma), TBF-beta/SMAD4 pathway (colon and rectum), cell cycle pathway (low-grade glioma). Thus, genes with third-order interactions are indeed enriched for shared molecular functions and pathways. We suggest a model where, in some pathways, single mutations in two different genes can sometimes have very similar consequences as two different mutations in just one of the genes. The ‘second hit’ in a tumor is sometimes a second genetic alteration in the same gene and sometimes the first mutation in a different gene in the same control pathway.

Fig2. Mutations that switch genes from two-hit to one-hit drivers in six cancer types. Changes in the strength of interaction between mutations and copy-number alterations in driver genes (‘gene A’) in the absence or presence of mutations in a second cancer gene (‘gene B’). A summary of the pathway in which the genes are involved is also shown.
Our study suggests that higher-order interactions are important for understanding cancer genetics: not only do the effects of individual mutations and their pairwise interactions need to be considered, but also what happens when three or more alterations are combined. Finally, we think that, although our results apply to human cancers, it is likely that the principles about genetic architecture will also apply to other diseases and traits. In other words, in future work, systematic identification of higher-order genetic interactions may also help understand the molecular mechanisms that cause other human diseases.
On a personal note, working on this project during the pandemic period as an early-career researcher taking my first steps to independence and also as a mother with a young child was quite a challenging task. I needed to manage my time efficiently by splitting my time as a supervisor of my students in the new group and also as a first-author of this study. However, it was a great joy to work continuously with my (former) colleagues and count on their support by sharing all scientific and also personal concerns regularly even through remote meetings between Barcelona and Madrid. I look forward to collaborating with Fran and Ben on many future projects for a better understanding of cancer fitness landscapes.
References
- Knudson, A.G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68, 820-823 (1971).
- Kern, S.E. Whose hypothesis? Ciphering, sectorials, D lesions, freckles and the operation of Stigler's Law. Cancer Biol Ther 1, 571-581 (2002).
- Berger, A.H., Knudson, A.G. & Pandolfi, P.P. A continuum model for tumour suppression. Nature 476, 163-169 (2011).
- Sherr, C.J. Principles of tumor suppression. Cell 116, 235-246 (2004).
- Nikolaev, S., et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 5, 5690 (2014).
- Takano, T., et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol 23, 6829-6837 (2005).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069-1075 (2008).
- Davoli, T., et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948-962 (2013).
- Lindeboom, R.G., Supek, F. & Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48, 1112-1118 (2016).
- Solimini, N.L., et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science 337, 104-109 (2012).
- Park, S. & Lehner, B. Cancer type-dependent genetic interactions between cancer driver alterations indicate plasticity of epistasis across cell types. Mol Syst Biol 11, 824 (2015).
- Park, S., Supek, F. & Lehner, B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat Commun 9, 2601 (2018).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in