Highly-Accurate, Long-Read DNA Sequencing Improves Analysis of a Human Genome
Published in Bioengineering & Biotechnology

DNA sequencing technologies have enabled amazing progress in our understanding of genomes. High-accuracy short-read sequencing (e.g., Illumina, Ion Torrent) excels at characterizing small variants in populations, but has limited ability to link variants into haplotypes, identify larger structural variants, and assemble genomes de novo. These applications are better suited to long-read sequencing technologies (e.g., PacBio, Oxford Nanopore), which provide long-range information but have lower read accuracy.
Years of development of PacBio’s single molecule, real-time (SMRT) Sequencing technology [1] have increased read length to an average of 100 kb. As featured in our recent paper, with circularized DNA templates, this read length provides 5-10 consecutive passes of single molecules in the 10-20 kb size range. Each pass is about 90% accurate, but the separate passes can be integrated in software to a single 10-20 kb consensus “HiFi” read that averages 99.9% accuracy at 10 passes (Figure 1).
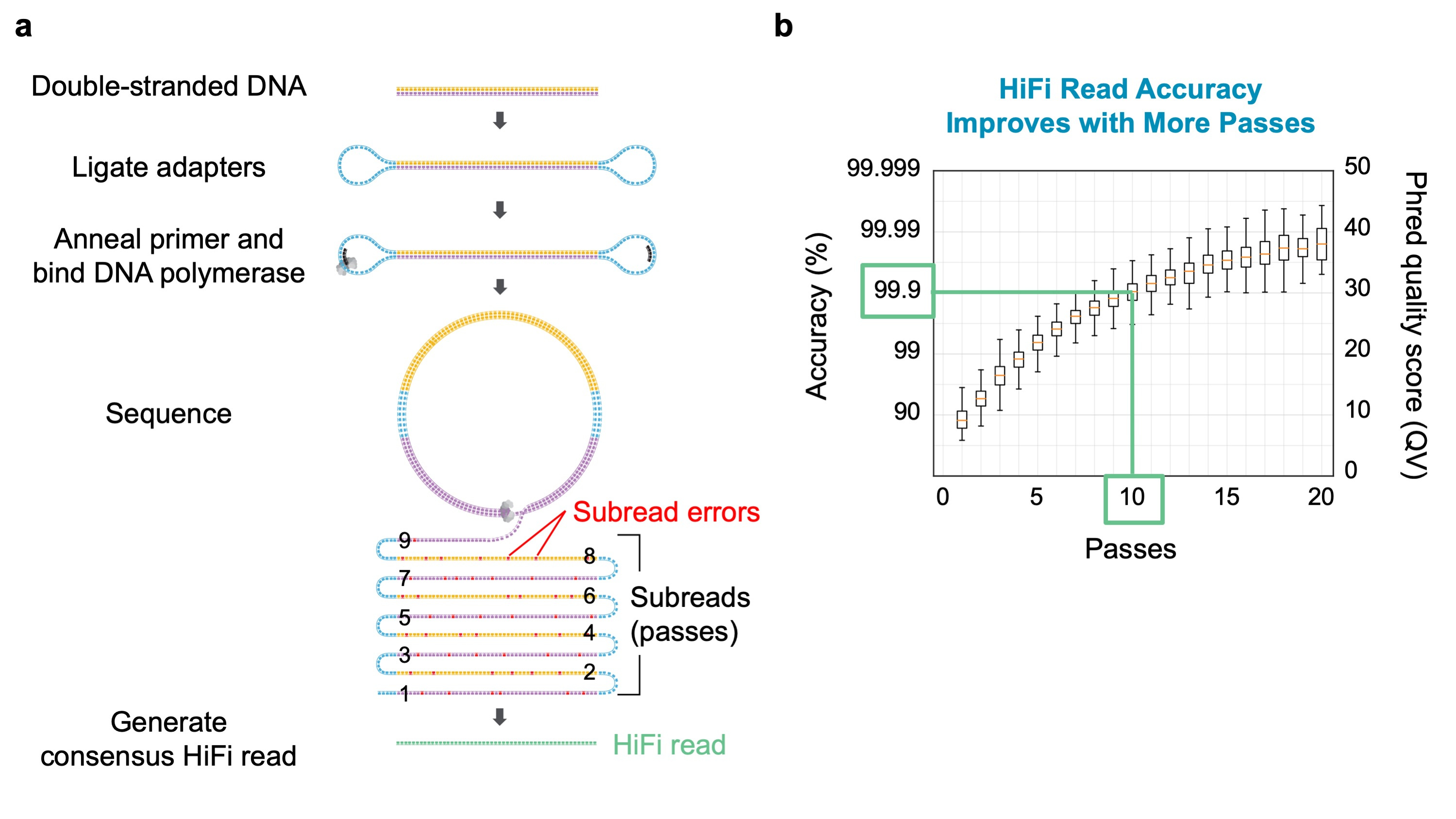
Figure 1. (a) Ligating hairpin adapters to a linear DNA template molecule produces a circular sequencing template. Long read length produces multiple forward and reverse strand reads which can be used to generate a highly accurate consensus sequence (HiFi read). (b)Consensus read accuracy as a function of the number of passes of the template molecule. In our study, we sequenced a genomic DNA library having insert molecules averaging 13.5 kb to a median of 10 passes, yielding HiFi reads with 99.9% accuracy.
To assess the value of these long and accurate HiFi reads, we needed to sequence a very well characterized genome. We selected the Genome in a Bottle human reference sample HG002 (NIST Reference Material 8391), which was evaluated with multiple sequencing technologies to develop a benchmark with an estimated accuracy of 1 error per 4 million basepairs within the defined benchmark regions [2]. We sequenced an HG002 genomic DNA library to a depth of 28-fold, obtaining HiFi reads averaging 13.5 kb, and examined our ability to identify variants and assemble the genome.
Using this HiFi data, Google DeepVariant[3] called single nucleotide variants better than industry-standard workflows that use short reads and the GATK pipeline (F1 score of 99.936% vs 99.881%). HiFi reads also increased the portion of the genome that was callable, and importantly extended coverage to 152 medically relevant genes that are not accessible with short-read techniques. For calling larger structural variants, the HiFi reads far outperformed other techniques, achieving F1 score of 96.06% compared to 67.53% for short reads. Furthermore, the structural variants were defined at an unprecedented base-level accuracy when compared with standard long-read techniques, which typically do not define breakpoints and lengths accurately. Lastly, de novoassemblies generated using HiFi data were of comparable contiguity (>25 Mb) to long-read assemblies but were much more accurate: 6-times more than polished PacBio-only assemblies, 80-times more than Oxford Nanopore assemblies polished with Illumina reads, and 200-times more than polished Oxford Nanopore-only assemblies.
HiFi reads provide both read length and accuracy, and thus are versatile for short- and long-range applications. Around 15-fold coverage of a human genome results in high precision and recall for SNVs, indels, and structural variants; and accurate, contiguous de novoassemblies. Generating that coverage for our publication required 20 SMRT Cells 1M and 23 days of sequencing on the PacBio Sequel System. The newer Sequel II System reduces that to 2 SMRT Cells 8M and 3 days of sequencing, making it accessible for individual labs to sequence a human genome with HiFi reads today. We expect that continued technology development will further improve cost and throughput.
REFERENCES
- Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science323, 133–138 (2009).
- Zook, J. M. et al.An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 37, 561-566 (2019).
- Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in