How we can simulate temporal changes of molecular networks based on the cause-result relationship?
Published in Cancer, Neuroscience, and General & Internal Medicine
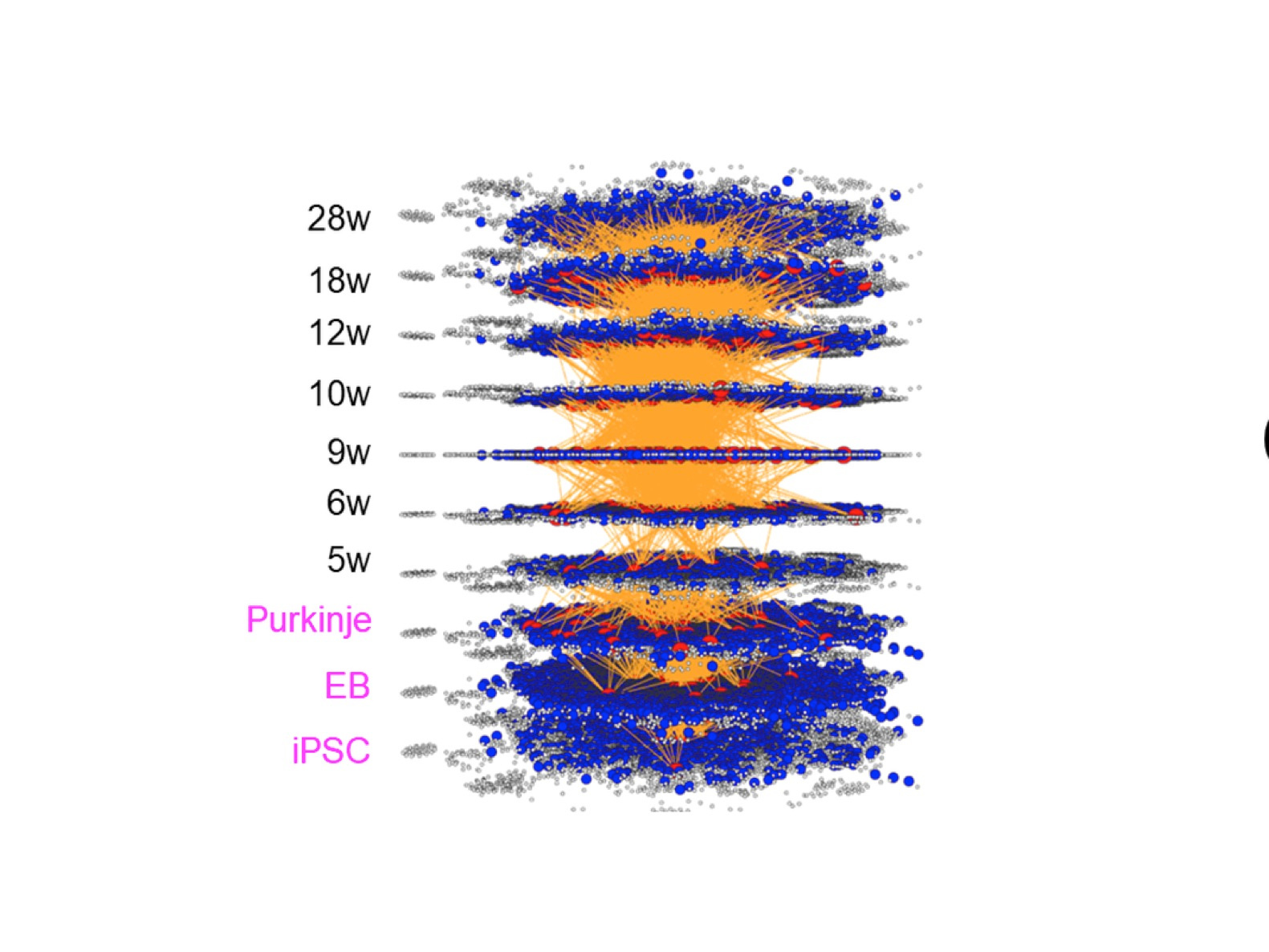
We are living in the four dimension (4D) world composed of space position (x, y, z) and time point. Molecular network in living cells, tissue, organ and organism is generally described in 2D while the reality is dynamically changed during time progression in 3D. Such a dynamic molecular network can be described with comprehensive omics data, especially by single cell RNA-seq or other single cell omics data in recent studies, while it remains difficult from which to extract the cause-result relationship and further prospect the future changes.
Naturally applying automatic intelligence (AI) to this issue is a promising idea whereas deep learning for such a huge molecular network demands an enormous size of observatory data. It is actually impossible for most laboratories in the world to obtain a necessary amount of data for their specific questions in biology. Therefore, we need some intermediate methods to overcome the difficulty.
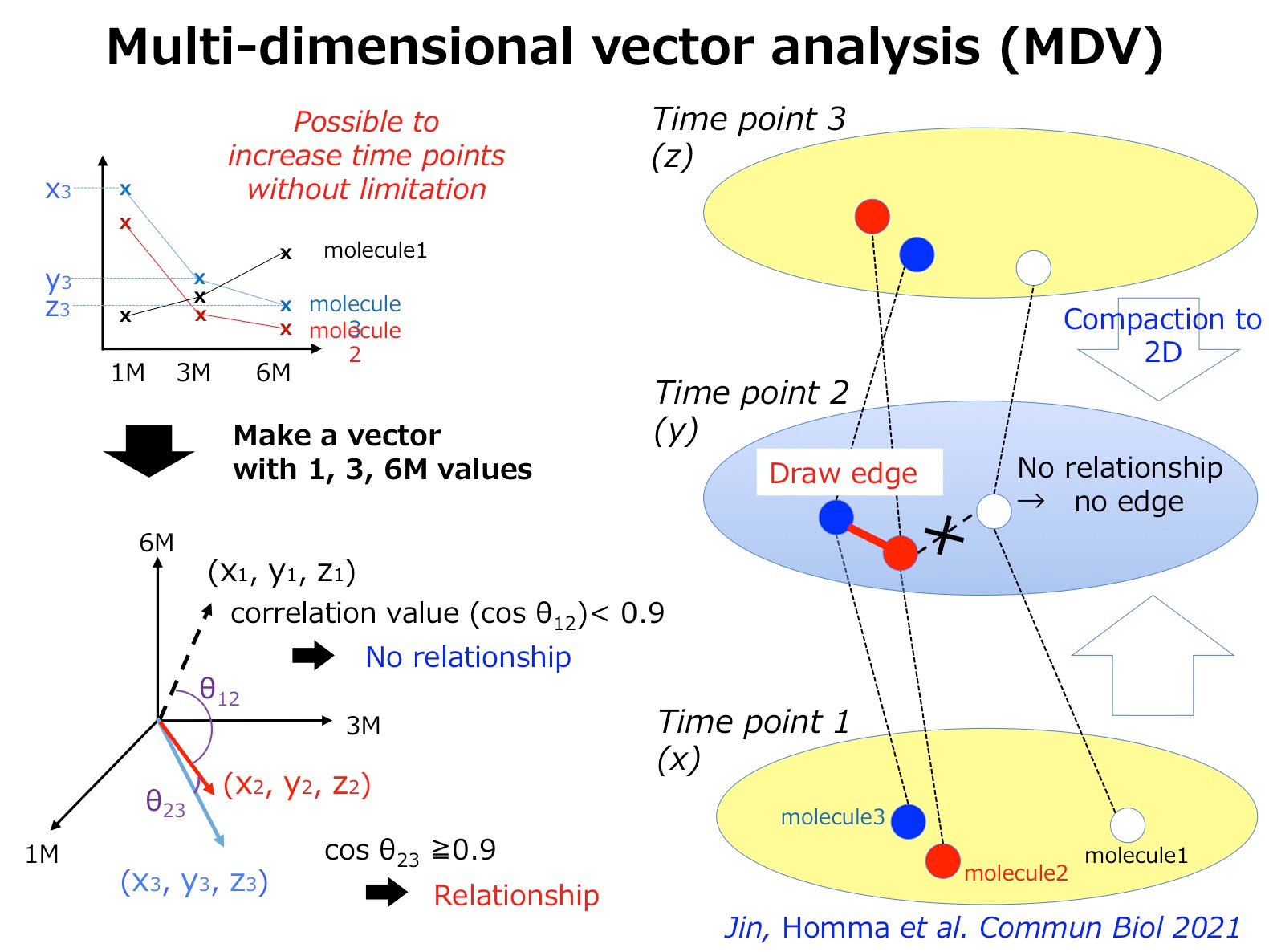
Two years ago, my group has published a method named multi-dimensional vector analysis (MDV) previously to analyze dynamic molecular networks in Alzheimer’s disease (AD) and fromtotempral lobar degeneration (FTLD), by which we could reveal the common pathological networks shared by two types of dementias (1) (Figure 1). MDV results indicated that the cell signaling from HMGB1-TLR4 interaction lead to PKC-MEK-MAPK/ERK activation and induces neurodegeneration (1).
Figure 1
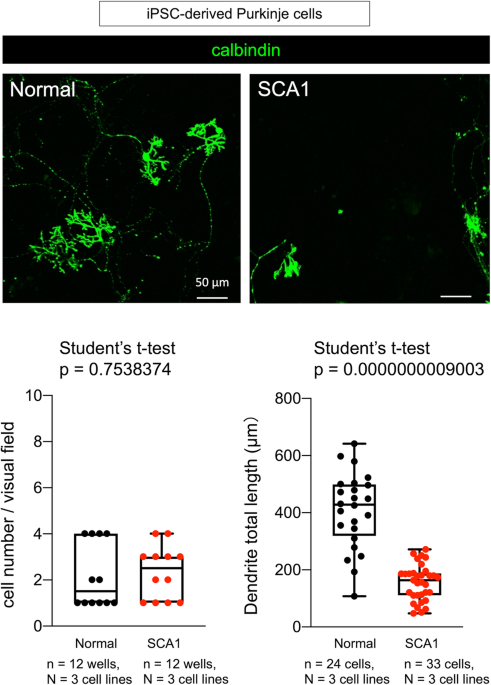
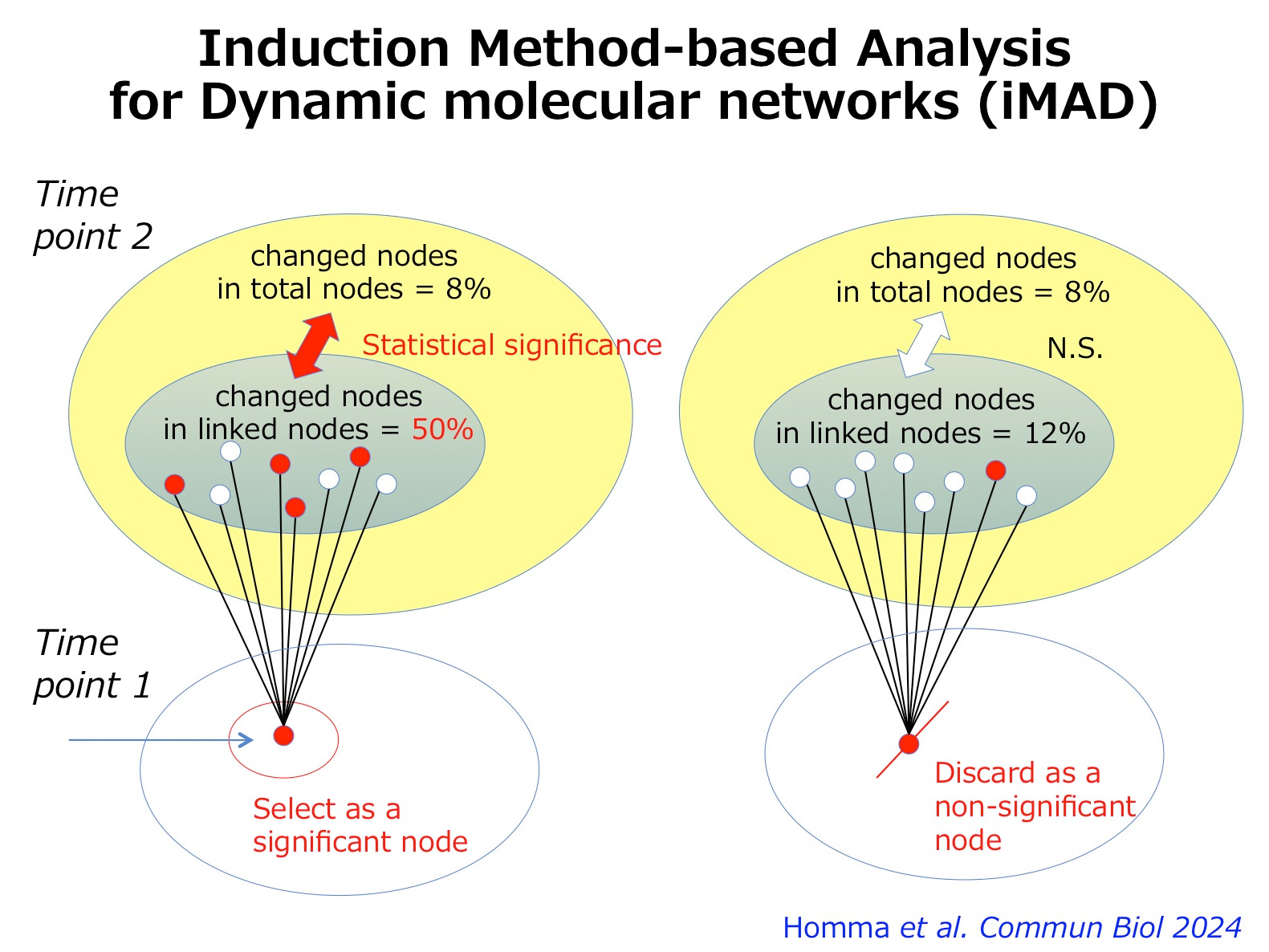
Now, we proposed another method named as induction method-based analysis for dynamic molecular networks (iMAD) (2) (Figure 2), and investigated the temporal pathology of spinocerebellar ataxia type 1 (SCA1) with our original RNA-seq data from human SCA1 patient-derived iPS cells and with public available data base from mouse SCA1 models (2). The iMAD analysis of cell differentiation from iPSC to Purkinje cells and of the meta-analysis by combining mouse data both uncovered unexpectedly early changes of immune response-related genes originated from the histone genes at the iPSC stage. Such prenatal changes cause the postnatal increase of ISG15 which influences ubiquitination of the disease protein Ataxin-1 and also brain inflammation at the early stage of adulthood (2).
Figure 2
Our two methods would be available for revealing previously unknown molecular changes that might be more critical than previously known changes of cell, tissue, organ and organism in various kinds of disease fields. We are preparing a commercial basis to provide the support of these data analyses for a wide range of scientists and companies who are interested in this approach. Please contact us (okazawa-tky@umin.ac.jp) if you are interested in collaboration.
References
1. Jin M, Jin X, Homma H, Fujita K, Tanaka H, Murayama S, Akatsu H, Tagawa K & Okazawa H. Prediction and verification of the AD-FTLD common pathomechanism based on dynamic molecular network analysis. Commun Biol. 4, 961 (2021). doi: 10.1038/s42003-021-02475-6.
2. Homma H, Yoshioka Y, Fujita K, Shirai S, Hama Y, Komano H, Saito Y, Yabe I, Okano H, Sasaki H, Tanaka H & Okazawa H. Dynamic molecular network analysis of iPSC-Purkinje cells differentiation delineates roles of ISG15 in SCA1 at the earliest stage. Commun Biol 7, 413 (2024). https://doi.org/10.1038/s42003-024-06066-z
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in