Hydrogenative alkene perdeuteration aided by a transient cooperative ligand
Published in Chemistry

Deuterium-enriched organic compounds have long been employed in chemical and biological isotopic labeling studies, and used commercially as solvents for nuclear magnetic resonance (NMR) spectroscopy. Attempts to introduce such compounds as drug candidates have also been made over the past half century, but only in 2017 was the first deuterated drug, deutetrabenazine (Fig. 1a), approved for therapeutic use by the United States Food and Drug Administration.1 This has bolstered interest in the deuteration of organic molecules as part of drug discovery processes, and many deuterated variants of existing pharmaceuticals, as well as entirely new compounds, are currently undergoing clinical trials2. These deuterated drug candidates, obtained through multistep syntheses involving expensive deuterium sources, typically feature perdeuterated variants of alkyl groups at specific sites, alongside undeuterated arene rings.

Fig. 1 | Deuterated drugs and hydrogenative perdeuteration of C=C bonds. a, Typical examples of deuterated drugs and candidates. b, Hydrogenative perdeuteration of C=C bonds using H2 and D2O in a single synthetic step. c, The transient cooperative thiol ligand balances two different reactions - D2 generation and C=C deuterogenation (r, reaction rate; red arrows pointing up and down represent rate increase and decrease, respectively).
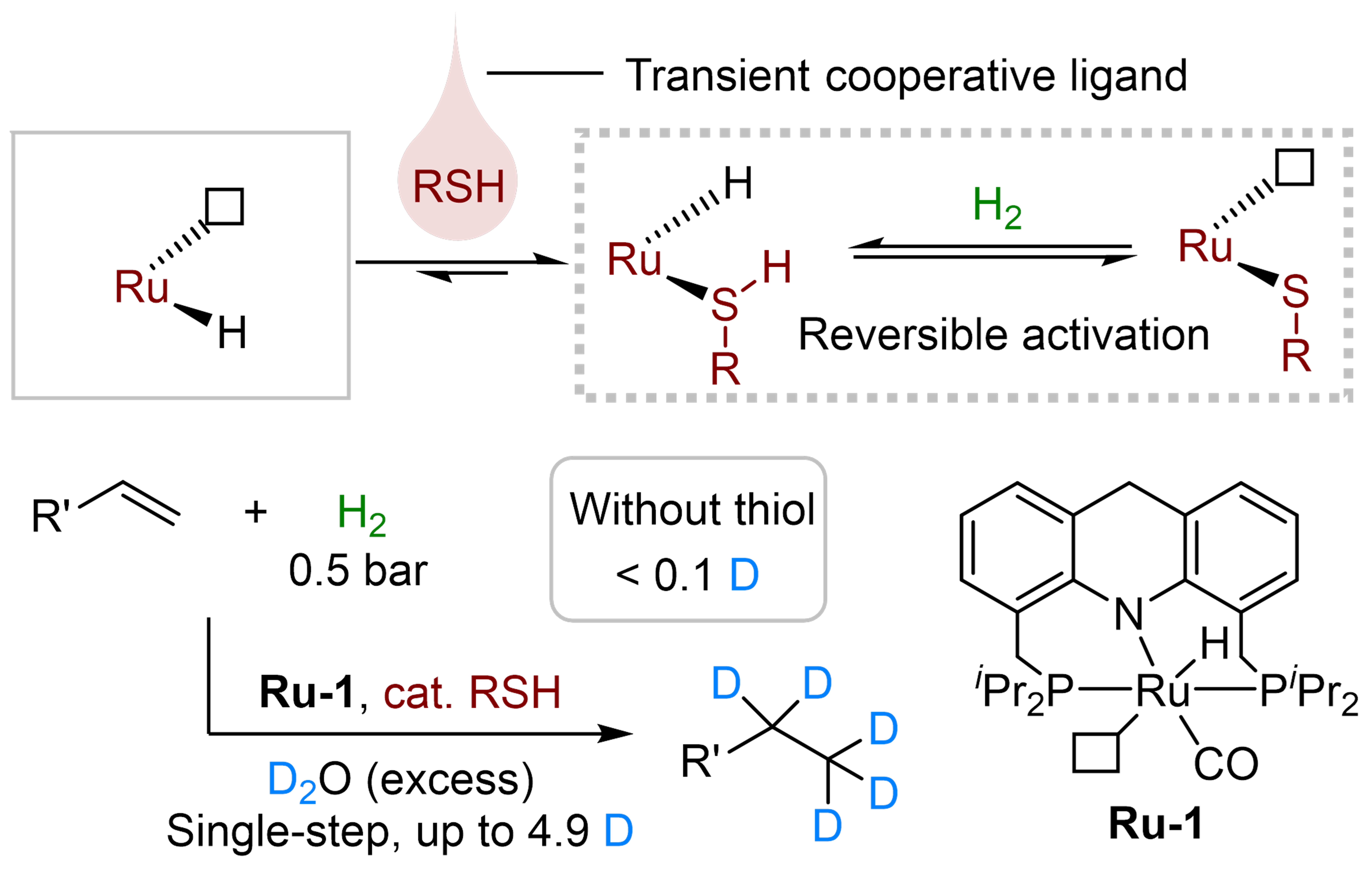
We post here our recently reported ruthenium-pincer-based catalytic system capable of performing an unprecedented hydrogenative perdeuteration of alkenes for the construction of perdeuterated alkyl groups at specific molecular sites, utilizing H2 and D2O in a single synthetic step, without the need for directly using expensive D2 gas (Fig. 1b). The success of this catalytic system is enabled by a catalytic amount of a thiol, which acts as a transient cooperative ligand (TCL), a concept we summarized in this work. The thiol not only accelerates the deuterium labeling of H2 with D2O through metal-ligand cooperation (rb, Fig. 1c),3 but also selectively inhibits the interaction of the catalyst with alkenes via its competitive coordination to the metal center (ra, Fig. 1c).
In detail, Ru-1 catalyzed H/D exchange of deuterated water (D2O) and H2 to generate D2 without using any sacrificial reagents or inputting electricity energy (Fig. 2a).4 Nevertheless, the deuterium labeling rate of this system was very slow with a TOF (H/D) of only 8 h-1. Interestingly, a catalytic amount of thiol greatly accelerates this H/D exchange rate by more than 20-folds (Fig. 2b), which is in line with the acceleration effect of metal-ligand cooperation that has been shown to efficiently enhance the reaction rate of the methanol reforming reaction.5 However, increasing the amounts of added thiol from 2.5 μmol to 10 μmol nearly didn’t inhibit this H/D exchange. This suggests that the reaction mechanism does not involve significant coordination of H2 or D2O to Ru-1, which otherwise would have become less coordinatively accessible as the concentration of thiol increases.
We’ve also applied this method of accelerated deuterium labeling of H2 with D2O to single-step deuterogenation of styrene 1a without using D2. Unexpectedly, over-deuterated D-2a, incorporating 3.4 deuterium atoms, was observed as the product using styrene 1a as a model substrate (Fig. 2c). Noteworthy, in the absence of thiol, nearly no deuterium incorporation was detected in the product 2a (<0.1 D incorporated). Moreover, increasing the amount of thiol enriched the isotope labeling in the product, albeit at the sacrifice of the product yields. The drop of the reaction rate indicates that the presence of thiol inhibits the reactions of styrene 1a in the system, which matches the inhibition effect and implies that the catalysis based on the original catalyst Ru-1 is involved in this alkene hydrogenative perdeuteration.6

Fig. 2 | Deuteration of H2 with D2O catalyzed by Ru-1. a, Reaction in the absence of thiol. b, Reaction in the presence of thiol. c, Inhibition effect of the relative amount of thiol.
The mechanism of the generation of perdeuterated D-2a involves reversible insertion of styrene into the Ru−D bond (Fig.3, r2),7 which occurs prior to the deuterogenation reaction (r3). Nevertheless, it’s worthy to mention a prerequisite for this reaction is that both D2 and the Ru−D species should be generated prior to hydrogenation (r1 > r3). Therefore, the result here reflects the significant transient cooperative role of the thiol during the reaction. Firstly, the added thiol accelerates the generation of Ru-deuteride species by cooperating with the ruthenium center. Secondly it selectively inhibits the reactions of the alkene by its competitive coordination to the metal center. These simultaneous acceleration and inhibition effects increase the rate difference between the generation of the Ru-deuteride species and its subsequent reactions with the alkene, thereby achieving the selective inclusion of deuterium at the double bond, rather than protium. This strategy, wherein the reaction rates of different tandem processes are balanced by using a transient cooperative ligand, is expected to be of further utility in the general field of catalysis.
 Fig. 3 | Thiol-accelerated H/D exchange and the competing thiol-inhibited olefin deuterogenation.
Fig. 3 | Thiol-accelerated H/D exchange and the competing thiol-inhibited olefin deuterogenation.
The developed system is effective for the hydrogenative perderuteration of various alkenes using H2 and D2O (Fig. 4). Up to 4.9 deuterium atoms could be incorporated into the products upon increasing the reaction temperature. Both aromatic and aliphatic substituted alkenes, as well as those with double bonds attached to heteroatoms, and even bioactive molecule derivatives could all be deuterogenated with abundant deuteriums selectively incorporated into the position of the double bond. Hopefully this method can contribute to the development of new deuterated drug candidates.

Fig. 4 | Substrate scope of hydrogenative perdeuteration of alkenes with H2 and D2O in a single step catalyzed by Ru-1 in the presence of catalytic thiol.
References
- Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493-494 (2017).
- Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276-5297 (2019).
- Khusnutdinova, J. R. & Milstein, D. Metal-ligand cooperation. Angew. Chem. Int. Ed. 54, 12236-12273 (2015).
- Sajiki, H. et al. Complete replacement of H2 by D2 via Pd/C-catalyzed H/D exchange reaction. Org. Lett. 6, 3521-3523 (2004).
- Luo, J. et al. Efficient base-free aqueous reforming of methanol homogeneously catalyzed by ruthenium exhibiting a remarkable acceleration by added catalytic thiol. J. Am. Chem. Soc. 143, 17284-17291 (2021).
- Luo, J. et al. Controlled selectivity through reversible inhibition of the catalyst: Stereodivergent semihydrogenation of alkynes. J. Am. Chem. Soc. 144, 13266-13275 (2022).
- Erdogan, G. & Grotjahn, D. B. Mild and selective deuteration and isomerization of alkenes by a bifunctional catalyst and deuterium oxide. J. Am. Chem. Soc. 131, 10354-10355 (2009).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in