Induced Pluripotent Stem Cells as an “off-the-shelf” Supply of T cells
Published in Bioengineering & Biotechnology

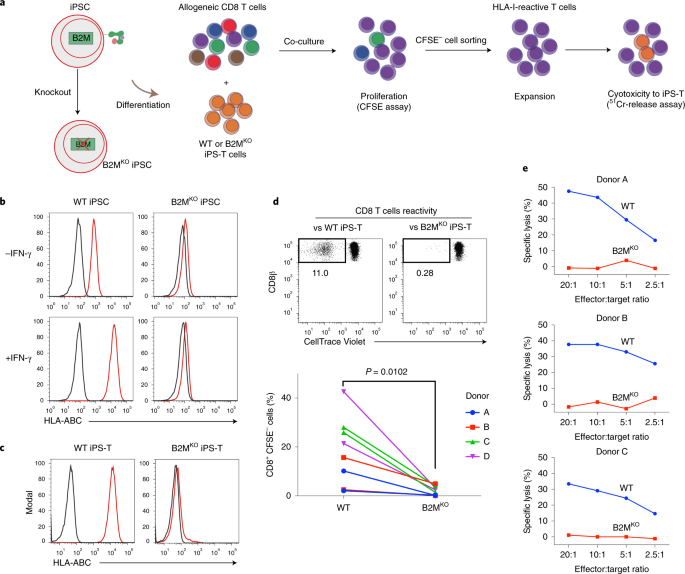
Autologous primary T cells engineered to express antigen-specific T cell receptors (TCRs) or chimeric antigen receptors (CARs) have great promise for clinical use against malignancies. However, the lengthy production process and high cost limit their large-scale clinical application. Using pre-banked allogenic T cells can circumvent these problems, but the immune rejection of transplanted allogenic T cells presents one major barrier. A recipient's CD8 T cells directly reject the grafted T cells by recognizing alloantigens (HLA class I or their presented antigens). Further, the recipient's CD4 T cells trigger the immune rejection of grafted T cells by recognizing other alloantigens (HLA class II or their presented antigens). Finally, the recipient's nature killer (NK) cells directly target grafted T cells that have a low expression of NK cell inhibitory ligands (normally HLA class I) and/or an overexpression of NK cell activating ligands. The regulation of these allogeneic factors and of factors that activate immune cells in T cells, mainly through gene modifications, should prevent immune rejection (Fig. 1) . However, multiple genetic modifications in primary T cells may lead to poor expansion and exhaustion.
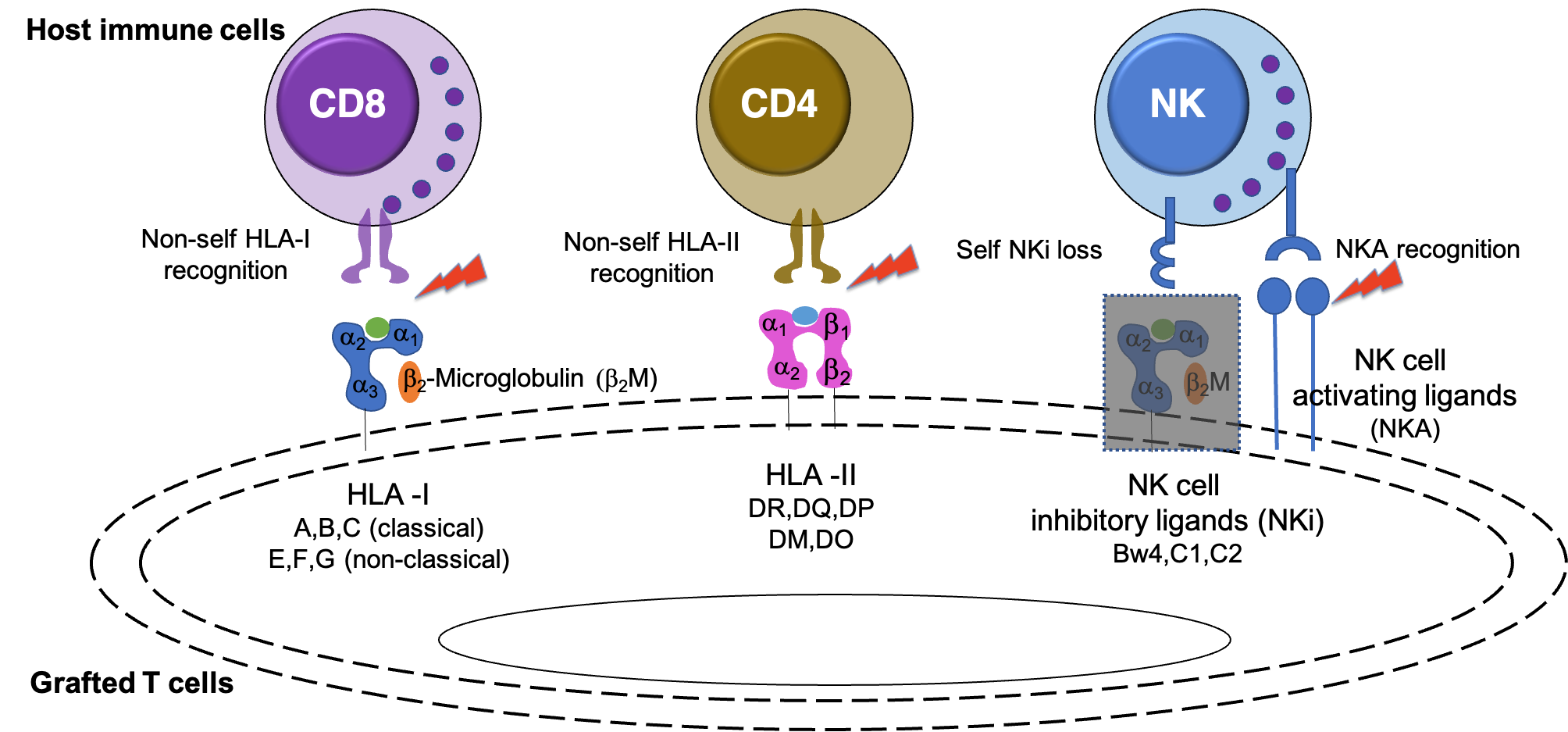
Figure 1. Target recognition during immune rejection.
We attempted to overcome these concerns by manipulating induced pluripotent stem cells (iPSCs), which can be maintained in an undifferentiated state before differentiation into mature T-cells. Gene modifications were performed in multiple steps in the clonal expandable iPSC stage. The iPSCs were then differentiated into T cells (iPS-T cells) and checked for immunogenicity.
In detail, we first checked that iPS-T cells naturally expressed HLA class I and upregulated HLA class II expression when activated. Accordingly, these iPS-T cells stimulated allogenic CD8 and CD4 T cells. To evade this stimulation, we generated HLA class I null iPSCs (B2MKO) by targeting the B2M gene, which encodes b2 microglobulin, a protein required for HLA class I presentation on the cell surface. We confirmed that B2MKO iPS-T cells do not induce the proliferation of allogenic CD8 T cells and escaped lysis. Next, we generated HLA class II null iPSCs (dKO) by knocking out the MHC class II transactivator CIITA in B2MKO iPSCs. Along with the B2MKO iPS-T cell effects on CD8 T cells, dKO iPS-T cells did not induce the activation or proliferation of allogenic CD4 T cells. However, HLA class I molecules also serve as major ligand inhibitors to NK cells. Indeed, we found that B2MKO and dKO iPS-T cells induced NK cell activation. NK cell activity is thought to be controlled by the balance of inhibitory and activating signals delivered via NK cell-surface receptors1. Conceivably then, increasing the level of ligands for inhibitory cell receptors on iPS-T cells or eliminating the ligands for NK cell activation receptors could abrogate the NK cell activation caused by B2MKO and dKO iPS-T cells. HLA-E (a ligand for the NK cell inhibitor receptor NKG2A) was reported to protect HLA class I down-regulated tissues, such as the placenta, and the overexpression of single chain HLA-E (B2M-HLA-E) protected B2MKO iPSC-derived CD45+ cells from NK cell lysis2. We therefore overexpressed HLA-E in dKO iPSCs (dKO/E iPSCs). dKO/E iPS-T cells caused low NKG2A+ NK cell activity. However, NKG2A- NK cells were still activated. To further control the NK cell activity, we eliminated PVR (a ligand for the NK cell activating receptor DNAM-1), which was upregulated in dKO/E iPS-T cells. These tKO/E iPS-T cells decreased the activity of DNAM-1+ NK cells, which is a population that includes both NKG2A+ and NKG2A– NK cells (Fig. 2).
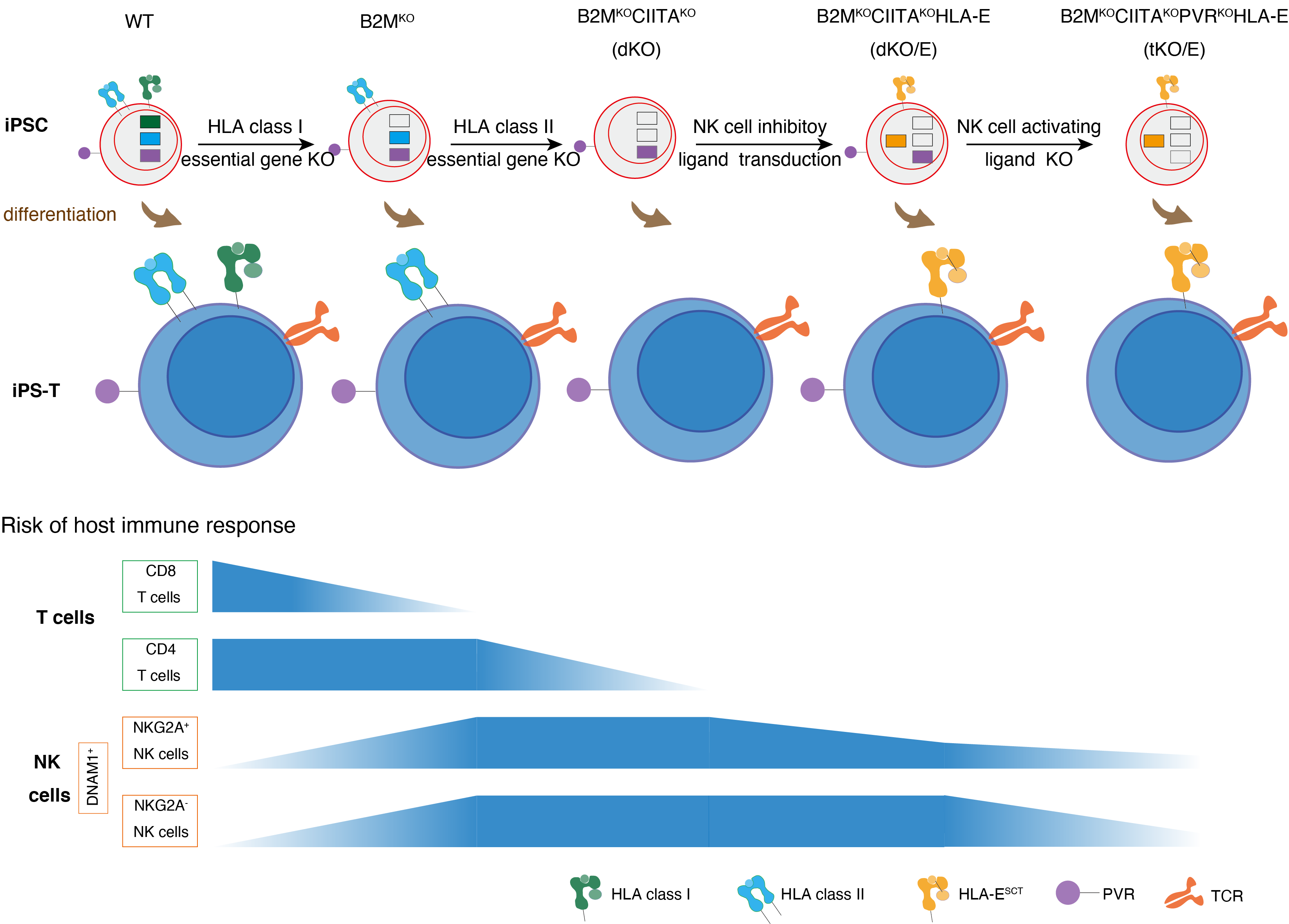
Figure 2. The strategy to generate hypoimmunogenic iPS-T cells.
Next, to clarify whether tKO/E iPS-T cells could functionally control tumor progression in the presence of alloreactive cells in a mouse model, we generated CD20+ immortalized B lymphocytes and co-transplanted these cells with their parent PBMCs into mice. Then, multiple doses of CD20-CAR tKO/E iPS-T cells were transplanted. Compared with CD20-CAR iPS-T cells, CD20-CAR tKO/E iPS-T cells showed longer survival in the presence of alloreactive cells, including CD4, CD8 T cells and NK cells, and were able to suppress the growth of immortalized B lymphocytes in mice.
Our research suggest that hypoimmunogenic iPSC-derived T cells may contribute to the creation of “off-the-shelf” T cell immunotherapies.
References:
1. López-Botet, M., Bellón, T. Natural killer cell activation and inhibition by receptors for MHC class I. Curr Opin Immunol 11, 301-307 (1999). doi:10.1016/s0952-7915(99)80048-x
2. Gornalusse, G., Hirata, R., Funk, S. et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 35, 765–772
Our paper: Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nature Biomedical Engineering (2021).
https://dx.doi.org/10.1038/s41551-021-00730-z
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Medical Ultrasound: Emerging Techniques and Applications
Publishing Model: Hybrid
Deadline: Jan 29, 2027




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in