Inferring patient-specific gene networks from bulk transcriptomics data
Published in Cancer, Protocols & Methods, and Genetics & Genomics
What are single-sample network inference tools and why do we need them?
Gene regulatory network inference has long relied on large sample sizes to robustly estimate statistical dependencies between genes from bulk transcriptomics data. The resulting networks are therefore population-level networks, in which individual features are averaged out. Yet, complex diseases such as cancer are characterized by a high inter-patient heterogeneity, and different disease subtypes require different treatment approaches. So how can we build patient-specific networks? Researchers can either turn to single-cell transcriptomics data, or find novel, clever ways to estimate single-sample networks from the large cohorts of bulk transcriptomics data out there, such as TCGA (The Cancer Genome Atlas Program) and CCLE (Cancer Cell Line Encyclopedia). Several of these methods make use of an aggregate network constructed from bulk all samples and a statistical wrapper to infer single-sample features within these networks. Others devise a specific statistic to directly obtain single-sample networks. Hence, single-sample network inference provides a welcome addition to a bioinformatician’s toolbox.
Promising as these tools seemed, we quickly noticed that deciding which tool to use in our own research projects was not obvious. A general, neutral benchmark or comparison of single-sample network inference tools was lacking, and each tool was presented using a different use-case in its original publication. Moreover, many of the compared methods rely on ‘normal tissue’ reference samples to contrast the tumour samples, which might not be available for all tumour types or in all precision oncology cases.
Comparing and evaluating the different tools
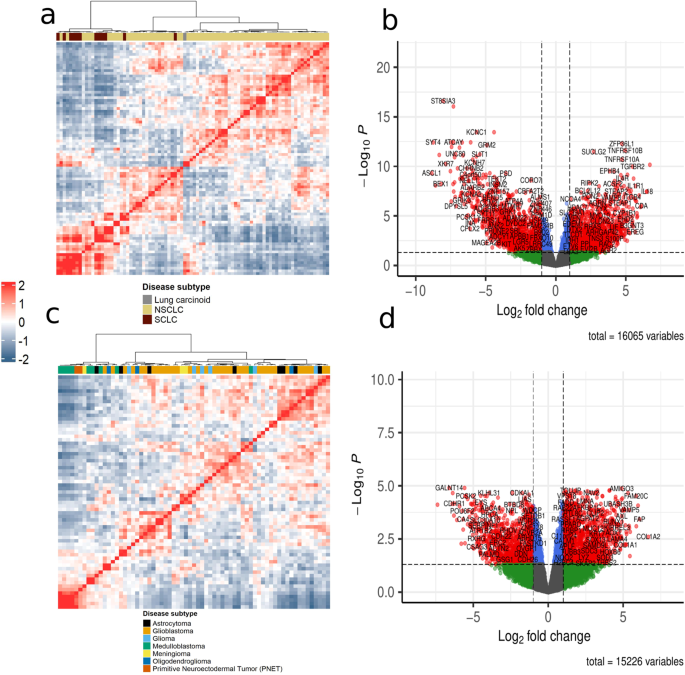
As there is no real ground-truth or gold-standard sample-specific dataset available to validate the predicted single-sample networks against, the comparative evaluation of the different methods was not straightforward. We tried to circumvent the absence of ground truth data in several ways, most importantly through exploiting other omics data generated on the same samples and by comparing to the aggregate network over all samples at population-level. Hence, we turned to the CCLE database, which in addition to cell lines’ transcriptomics data also contains other omics data such as proteomics, metabolomics and copy number variation data.
Next, we needed to investigate which tools to include in our study and how to include them. What kind of input data do they require, and what kind of output do we require to make them comparable? We decided to only include tools that allow to estimate single-sample networks without the need of a matched ‘healthy’ or reference sample, since these are often not available in clinical settings. Also, we required the tools to produce an output in network-like format, instead of e.g. a list of dysregulated pathways as estimated by ‘N-of-1- pathways’ or an activity score per gene such as inferred by VIPER. Moreover, the tools also needed to have their code publicly accessible. These criteria resulted in the selection of six tools, SSN, LIONESS, iENA, SWEET, CSN and SSPGI, which we slightly adapted to make them fully compatible in the evaluation.
We made a selection of 86 lung cancer and 67 brain cancer cell lines, each one additionally annotated with a cancer subtype and sub_subtype. Single-sample networks were constructed for each of these samples and we found that SSPGI was not scalable and that CSN networks had binary weights, which made ranking edges impossible. We also found that the algorithmically similar methods SSN and LIONESS resulted in structurally highly similar networks.
The main question we had was if these single-sample network inference methods provide more sample-specific information than the aggregate networks traditionally used on bulk transcriptomics data. Furthermore, we also investigated which tool would result in the most sample-specificity. For this we strongly relied on the cell lines’ cancer subtype and sub_subtype annotations as provided by CCLE. We hypothesized that hubs in the single-sample networks are related to the cancer subtype of a given sample. We found that SSN, LIONESS and iENA identified the largest part of subtype-specific hubs and hubs in these networks also differed the most from hubs in the aggregate network. We also hypothesized that known cancer driver genes for a specific cancer subtype should pop up in hub gene analysis or differential node importance analysis when comparing single-sample networks for a given set of cancer subtypes. Indeed, we found that hubs displayed enrichment for subtype-specific IntOGen/COSMIC drivers for NSCLC and glioblastoma, the two largest sample groups in respectively lung and brain samples. Next, we focused on additional datasets available in CCLE, coupled proteomics and copy number variation (CNV) data for our selection of cell lines. We evaluated the strength of correlation between the importance of a given gene in a single-sample network and proteomics or CNV measurement for the same gene in that sample. We were excited to observe stronger correlations for single-sample networks compared to the population-level networks in which individual characteristics are averaged out. Moreover, this approach allowed us to distinguish between methods, since SSN, LIONESS and SWEET generally outperformed the other tools. For SSN, LIONESS and iENA, it is important to balance different sample groups within the samples under study, since we observed a tendency for higher edge weights in the underrepresented sample group for these methods. SWEET includes a weighting factor to counteract this, but we found this to result in highly similar single-sample networks, albeit with more sample-specific information than the aggregate network.
Single-sample networks inference can contribute to precision oncology
Our results show that single-sample network inference algorithms can successfully be applied to prioritize molecular characteristics of individual samples or patients, which often reflect the underlying disease subtype. These tools are applied to bulk transcriptomics and without the need for a matching ‘healthy’ sample, allowing to mine the plethora of existing datasets or potentially allowing cost-effective implementation in clinical settings. We conclude that single-sample network inference methods can reflect sample-specific biology better than traditional bulk network inference when ‘normal tissue’ samples are absent and that each method has it own peculiarities.
Follow the Topic
-
npj Systems Biology and Applications
An online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach and encourages studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems.

Related Collections
With Collections, you can get published faster and increase your visibility.
Mechanistic and data-driven modeling in mathematical oncology
Publishing Model: Open Access
Deadline: Dec 02, 2026
Systems mechanobiology
Publishing Model: Open Access
Deadline: Sep 28, 2026





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in