Inflammation remodels the immune microenvironment in acute myeloid leukemia
Published in Cancer
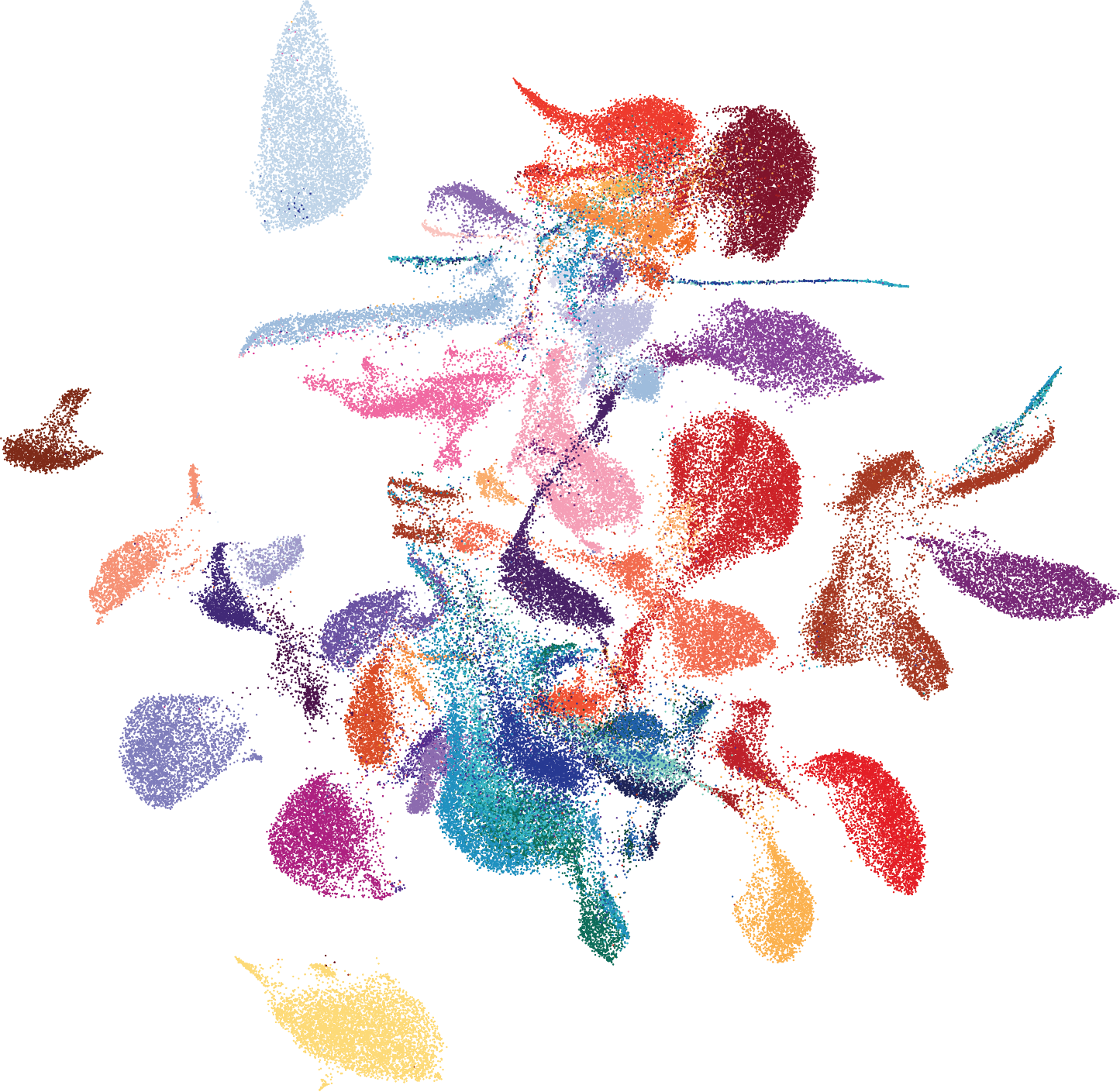

Acute myeloid leukemia (AML) is an aggressive blood cancer with poor prognosis and limited treatment options. Although new targeted therapies have been approved in recent years, survival is still dismal. In our work, we set out to characterize the immune microenvironment in AML to identify potential novel avenues for treatment.
Inflammation is a hallmark of cancer and is known to affect disease progression and outcome in many different types of tumors. In myeloid cancers, inflammation has been linked to very early stages of the disease, starting with a pre-malignant condition known as clonal hematopoiesis (CH), where blood cells carrying specific AML-related mutations accumulate in the blood. Inflammation is also implicated in the transition from an AML-precursor disease, myelodysplastic syndrome, to overt AML. However, the role of inflammation in shaping the immune microenvironment of AML remains unclear.
To study the immune microenvironment in AML, we performed single-cell RNA-sequencing (scRNA-Seq) combined with profiling of cell surface proteins (CITE-Seq) on diagnostic bone marrow aspirates from healthy donors, adult AML patients, and pediatric AML patients. We characterized all cell lineages in the bone marrow based on RNA and cell surface protein expression. We separated malignant and microenvironment cells based on identification of chromosomal gains or losses and patient-specific cell clustering. This thorough annotation allowed us to further delve into the transcriptional programs unique to malignant cells, and how they affect the immune microenvironment. Analysis of malignant cells revealed that inflammation is a common pathogenic module expressed by tumor cells in both adult and pediatric patients. Further examination of differentially-expressed genes between malignant cells and their healthy counterparts allowed us to generate adult- and pediatric-specific inflammation gene signatures, which we then used to divide the patients into high- and low-inflammation groups and compare their immune microenvironment.
Our analysis of the immune microenvironment identified several differences between high and low inflammation patients. A suppressive B cell population, atypical memory B cells, was expanded in high inflammation patients. These cells were previously identified in patients with chronic or recurrent infections, such as malaria, HIV and hepatitis C virus, as well as in patients with severe COVID-19, and are thought to have a lower capacity to secrete antibodies against these pathogens. In line with this, atypical B cells from AML patients expressed genes associated with B cell dysfunction. Notably, atypical B cells correlated with high inflammation in several additional cohorts of AML patients. In pediatric AML patients, we additionally identified inflammation-related changes in the T cell compartment. High-inflammation patients had higher levels of a suppressive T cell population, regulatory T cells, and progenitor exhausted T cells. Progenitor exhausted T cells are thought to preferentially respond to treatment with immune checkpoint blockade, suggesting that high-inflammation pediatric patients may benefit from this treatment, which is not currently offered to AML patients. We also examined the effect of inflammation on T cell clonal expansion, and demonstrated that in patients with high inflammation T cell clonal expansion is reduced, suggesting that the T cell response in these patients is less active.
We then turned to examine T cell clonal expansion in adult and pediatric patients. While adult patients presented with clonally expanded CD8+ T cells, infant patients did not show clonal T cell expansion. In infants, AML is thought to develop during fetal development, and it is possible that early acquisition of mutations induces tolerance and blunts the T cell response to AML.
Finally, we examined the effect of inflammation on patient survival. We identified 38 inflammation-related genes in adults and 11 inflammation-related genes in pediatric patients and generated a clinically-relevant inflammation risk score (iScore). In AML, patients are stratified into risk groups based on the mutations that their tumors carry, their age and other clinical parameters. Treatment is then determined by risk status, where patients with higher risk receive more intensive treatment and bone marrow transplants. We found that incorporation of the iScore into this risk stratification strategy can improve overall risk stratification, enabling the identification of patients that will benefit from more intensive treatment. Notably, for patients currently classified as low risk, high iScore confers a similar outcome to high-risk patients, suggesting this group will particularly benefit from clinical incorporation of the iScore. In addition, we found that high iScore associates with specific mutations in both adult and pediatric patients, enabling identification of patients at higher risk.
This study provides the first detailed analysis of the immune microenvironment in AML, as well as the first description of the effect of inflammation on the AML immune microenvironment. We hope that our findings will be implemented in the clinic, and lead to improved outcomes for AML patients.
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in