Insights on genomic prediction and underlying molecular mechanisms for complex traits by integrating multi-omics data
Published in Genetics & Genomics
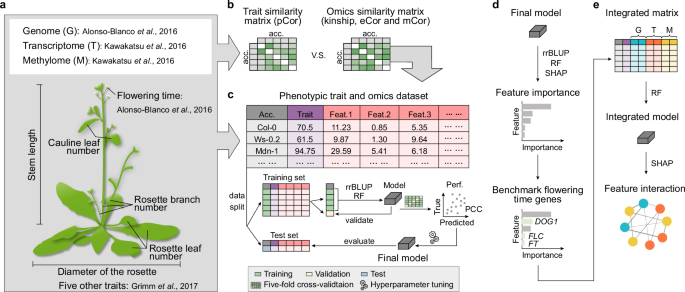
Plant complex traits are determined by the genotype, environments and the genotype-by-environment interactions. Regarding the genotype, beyond the genome sequences, the complex traits are also influenced by the composition and abundance of transcripts and proteins, the DNA methylation, chromosome modifications and 3D chromosome structures, and so on. The Arabidopsis 1001 Genome Project has generated the genomic (G), transcriptomic (T), methylomic (M) and six phenomic data for hundreds of Arabidopsis accessions1–3, which were planted under the same environmental conditions (Fig. 1). These resources allow us to assess the contribution of three types of gene-based features to the prediction of complex traits and uncover potential interactions between these three omics data4.
Figure 1. Multi-omics data in Arabidopsis
Although the phenotypic similarities among accessions were not well reflected by genetic similarities among accessions, regardless of the type of omics data used, three flowering time related traits—flowering time, rosette leaf number, and cauline leaf number—can be predicted with higher accuracy when these three types of omics data were used than when the population structure was used. The transcriptome and methylome data-based models had comparable prediction accuracy as models built using genome data that is commonly used by the community. In addition, prediction accuracy of models built using methylomic data can be improved by considering single-site-based methylation beyond gene-body methylation (gbM).
By interpreting the predictive models, some benchmark flowering time genes were identified as important for predicting three flowering time-related traits. The identification of benchmark flowering time genes can be influenced by the types of omics data, the ways to represent data, the feature importance measuring approaches, the environmental conditions (e.g., environmental temperatures investigated in this study) under which the complex traits were measured, the accessions used to train the predictive models, and the ways that the benchmark genes were determined in previous studies. These results provided some caveats and guidelines for studies aiming at identifying potential contributing genes for plant complex traits using omics data. In addition, nine additional genes, that were important for predicting flowering time but had not been reported previously in regulating flowering time, were found to significantly alter flowering time when mutated.
The contribution of genetic features to complex trait prediction were accession-dependent, which was uncovered by examining the SHapley Additive exPlanations (SHAP)5 values of features for individual accessions in the predictive models. The decoupled expression and contribution to flowering time prediction between three hub genes in flowering time regulation network—SOC1, FT, and FLC—in some accessions (Fig. 2) suggests that knowledge for flowering time regulation in lab-used accessions may be not generalizable to other accessions. This finding raises a new question that how the flowering time regulation network evolved in the microevolutionary scale, which can be potentially answered by examining the evolutionary trajectory of genomic, transcriptomic and methylomic status for flowering time genes among accessions.
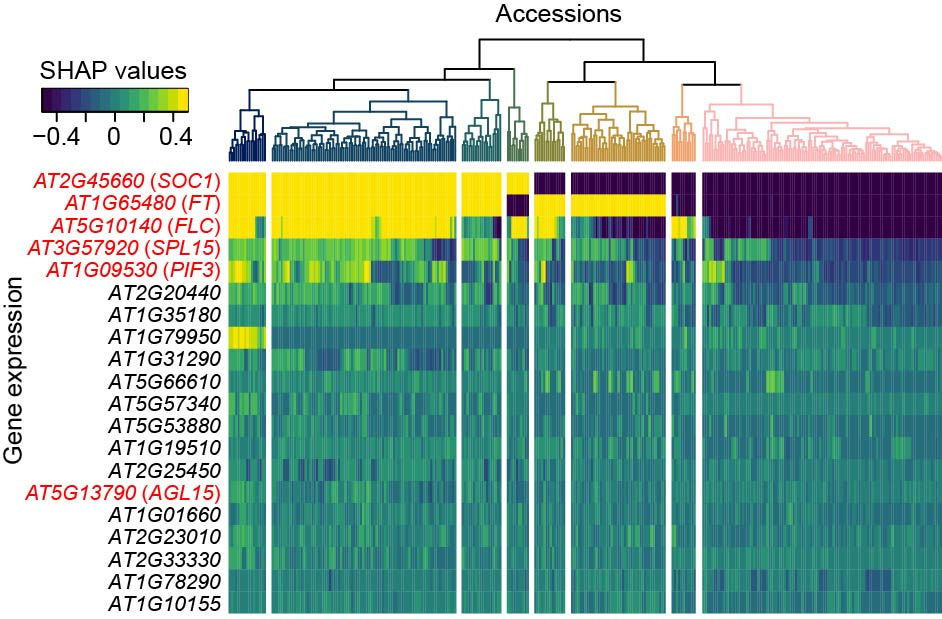
Figure 2. Accession-specific SHAP values of T-features
The flowering time prediction accuracy can be improved by integrating three types of omics data, potentially due to the inclusion of interactions among three omics data in the integrated models. The feature interaction network among three types of features of 426 benchmark flowering time genes simulated the reported flowering time regulatory network, as that the hub genes in the regulatory network were also hubs in the feature interaction network (Fig. 3). Beyond this, the feature interaction network revealed by interpreting the predictive models that integrated three omics data also provides insights on the potential genetic interactions among genes across three dimensions of genetic regulation.
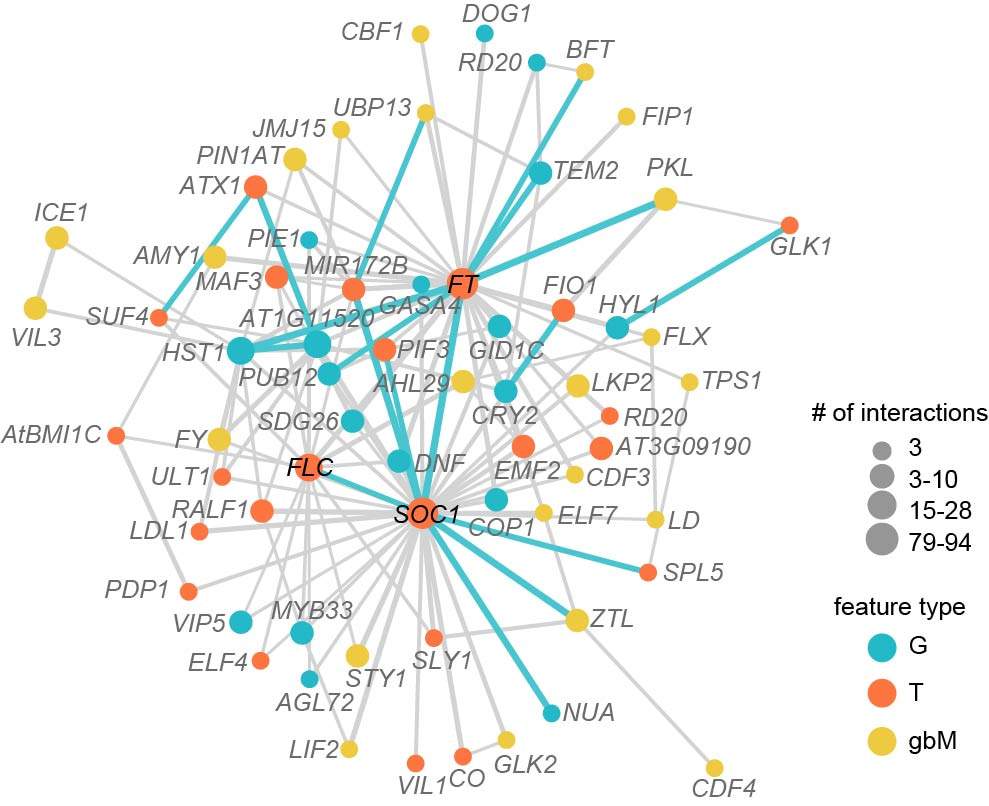
Figure 3. Feature interaction network among three types of features for benchmark flowering time genes
In summary, integrating multi-omics data in predicting plant complex traits can improve the prediction accuracy and extend our knowledge about the genetic mechanisms underlying plant complex traits.
This post is written by Peipei Wang (Kunpeng Institute of Modern Agriculture at Foshan, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences).
Reference
- Alonso-Blanco, C. et al. 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166, 481–491 (2016).
- Kawakatsu, T. et al. Epigenomic diversity in a global collection of Arabidopsis thalianaaccessions. Cell 166, 492–505 (2016).
- Grimm, D. G. et al. easyGWAS: A cloud-based platform for comparing the results of genome-wide association studies. Plant Cell 29, 5–19 (2017).
- Wang, P., Lehti-Shiu, M.D., Lotreck, S., Segura Abá, K., Krysan, P.J., Shiu, S.-H. Prediction of plant complex traits via integration of multi-omics data. Nat Commun 15(1):6856 (2024).
- Lundberg, S. & Lee, S.-I. A unified approach to interpreting model predictions. arXiv:1705.07874 [cs, stat] (2017).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in