Iron(III)-Based Metalloradical Catalysis for Asymmetric Cyclopropanation via a Stepwise Radical Mechanism
Published in Chemistry


One-electron radical chemistry has attracted growing attention in organic synthesis due to its distinct reactivity compared to traditional two-electron polar chemistry. Controlling the stereoselectivity, especially enantioselectivity, of radical reactions has long been a formidable challenge. Among recent advances, metalloradical catalysis (MRC) offers a conceptually different approach, achieving control over reactivity and stereoselectivity through the catalytic generation of metal-stabilized organic radicals as key intermediates governing subsequent homolytic radical reactions. Co(II) complexes of porphyrins ([Co(Por)])—stable 15e-metalloradicals—have been demonstrated as effective MRC catalysts, involving fundamentally distinct α-Co(III)-alkyl and α-Co(III)-aminyl radical intermediates. Introducing D2-symmetric chiral amidoporphyrins as supporting ligands, Co(II)-based MRC has successfully enabled various asymmetric radical reactions, including C=C bond cyclopropanation and aziridination, and C–H bond alkylation and amination, with precise control over reactivity and stereoselectivity. Expanding beyond Co(II)-based systems, we have explored other open-shell transition-metal complexes, in particular five-coordinate Fe(III) complexes of porphyrins ([Fe(Por)X]), as potential metalloradical catalysts for MRC. The low toxicity, abundance and cost-effectiveness of iron make Fe(III)-based metalloradical catalysts attractive, offering opportunities to explore diverse catalytic processes for stereoselective radical transformations.
Although the air-sensitive Fe(II) complexes of porphyrins have been studied extensively, stable Fe(III) complexes of porphyrins have not been unambiguously established as genuine catalysts for catalytic transformations in metallocarbene systems, especially in radical pathways. Although MRC has been primarily demonstrated using low-spin d7 [Co(Por)] (Fig. 1a), we questioned whether [Fe(Por)X], with the well-known spin-crossover of the d5 configuration, could also operate as potential metalloradical catalysts. Our focus was on investigating [Fe(Por)X] for olefin cyclopropanation with diazo compounds, a catalytic process previously presumed to proceed via in situ-reduced Fe(II) complexes and a concerted mechanism (Fig. 1b). We envisioned a stepwise radical mechanism for Fe(III)-catalysed cyclopropanation (Fig. 1c), generating trifluoromethyl-substituted cyclopropanes of great medicinal-chemistry interest (Supplementary Fig. 1). However, the ability of [Fe(Por)X] to homolytically activate diazo reagent 1’ and generate α-Fe(IV)-alkyl radical intermediate I remained unknown, along with crucial mechanistic considerations. Could α-Fe(IV)-alkyl radicals I serve as a catalytic intermediate for radical addition to substrate alkenes 2, and would the resulting γ-Fe(IV)-alkyl radicals II undergo intramolecular radical substitution via 3-exo-tet radical cyclization to deliver the cyclopropane products? Moreover, the diastereoselectivity and enantioselectivity controls of the radical transformation by Fe(III)-MRC have remained open questions. Drawing inspiration from Co(II)-MRC development, we anticipated D2-symmetric chiral amidoporphyrins to function as an effective ligand platform to support an Fe(III)-based metalloradical system and govern the stereochemical course of the proposed radical cyclopropanation.
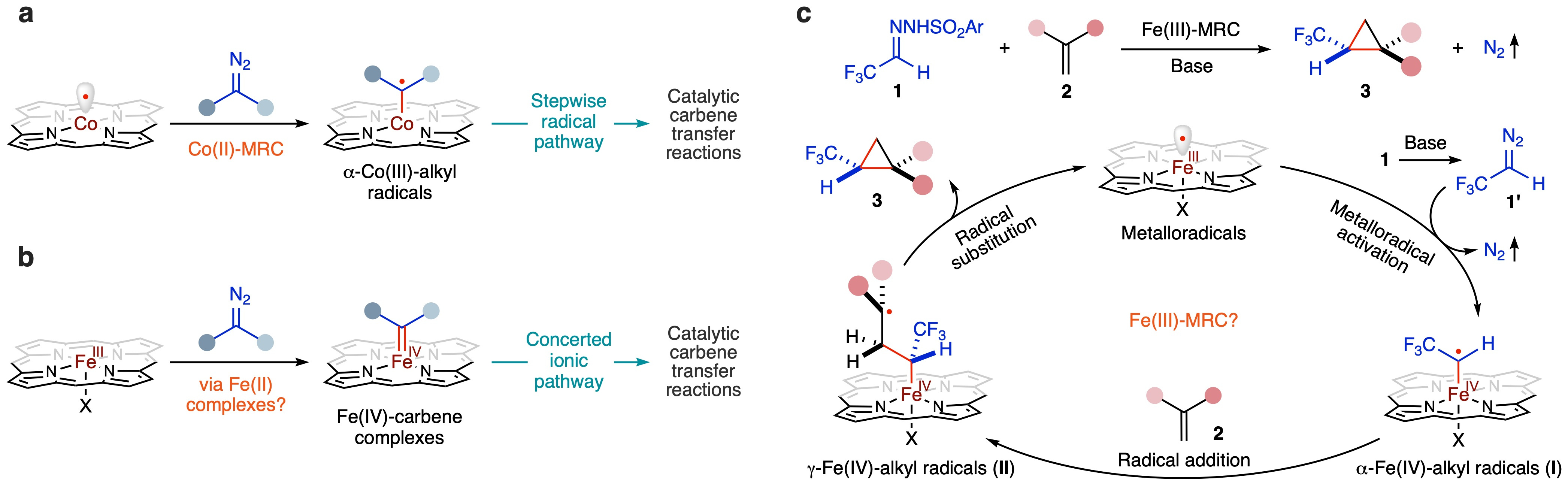
Fig. 1. Proposed stepwise radical mechanism for cyclopropanation of alkenes with in situ-generated α-trifluoromethyldiazomethane via Fe(III)-based metalloradical catalysis
Under optimized conditions, we evaluated the scope of [Fe(P3)Cl]-catalysed asymmetric cyclopropanation with in situ-generated trifluoromethyldiazomethane (1’) from hydrazone 1 in the presence of Cs2CO3, employing different alkenes as substrates (Table 1).
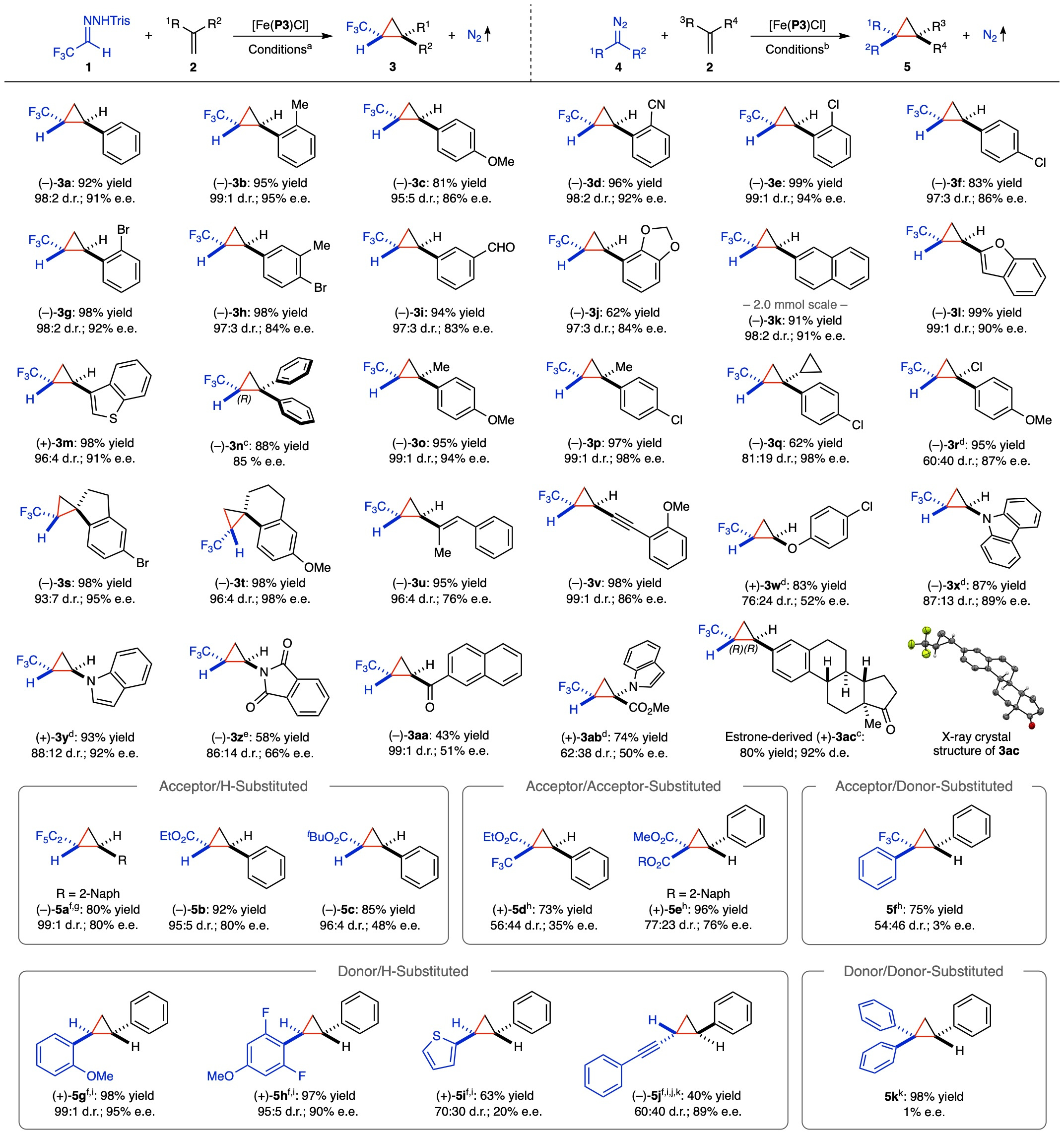
Table 1. Asymmetric cyclopropanation of alkenes by [Fe(P3)Cl] with in situ-generated α-trifluoromethyldiazomethane and various diazo compounds
To elucidate the underlying mechanism of the Fe(III)-based metalloradical system for olefin cyclopropanation, we performed experimental studies and density functional theory (DFT) computations.
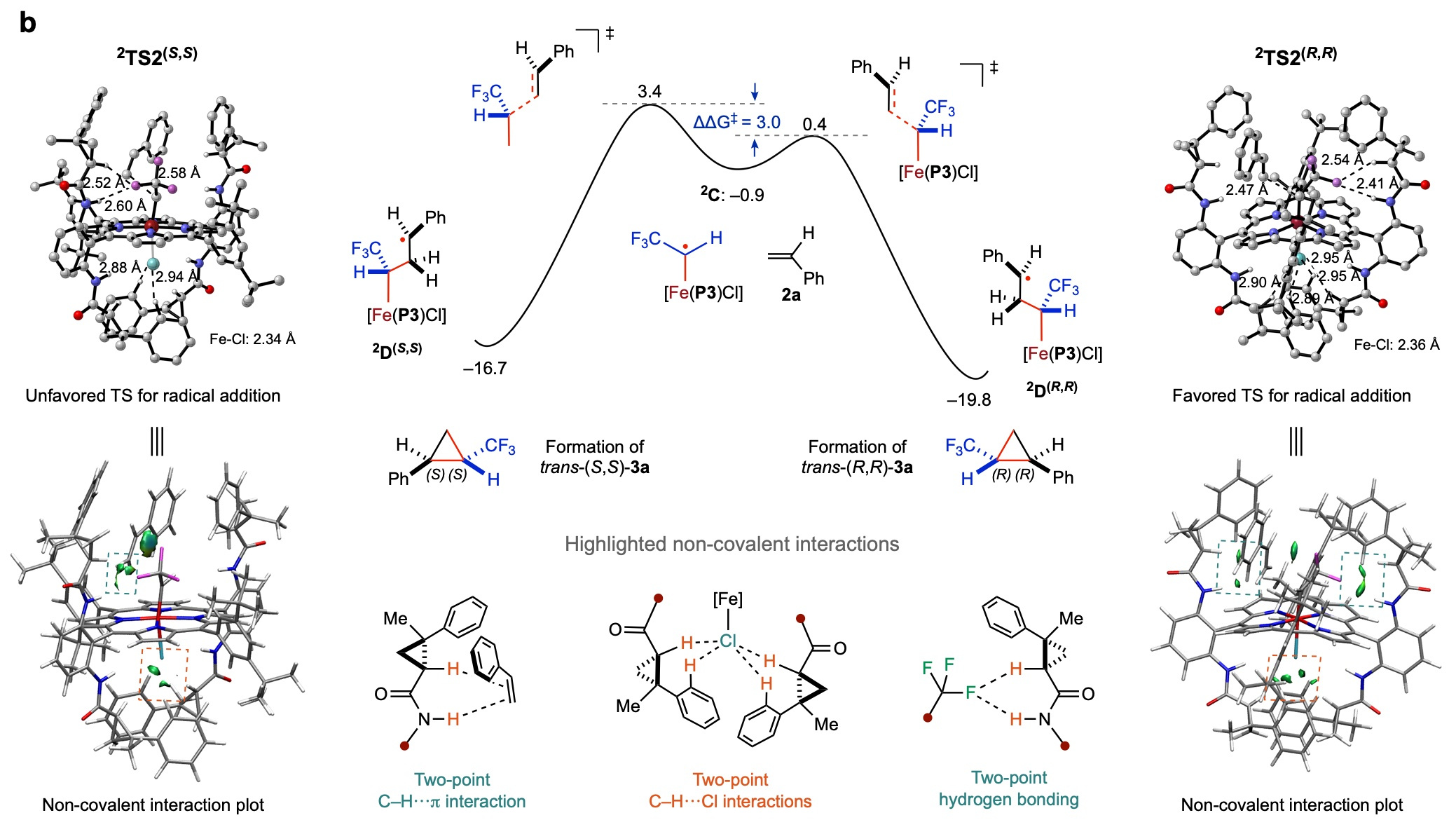
Fig. 2. DFT study on the origin of the enantioselectivity of the asymmetric cyclopropanation of styrene with trifluoromethyldiazomethane (1’) by [Fe(P3)Cl]
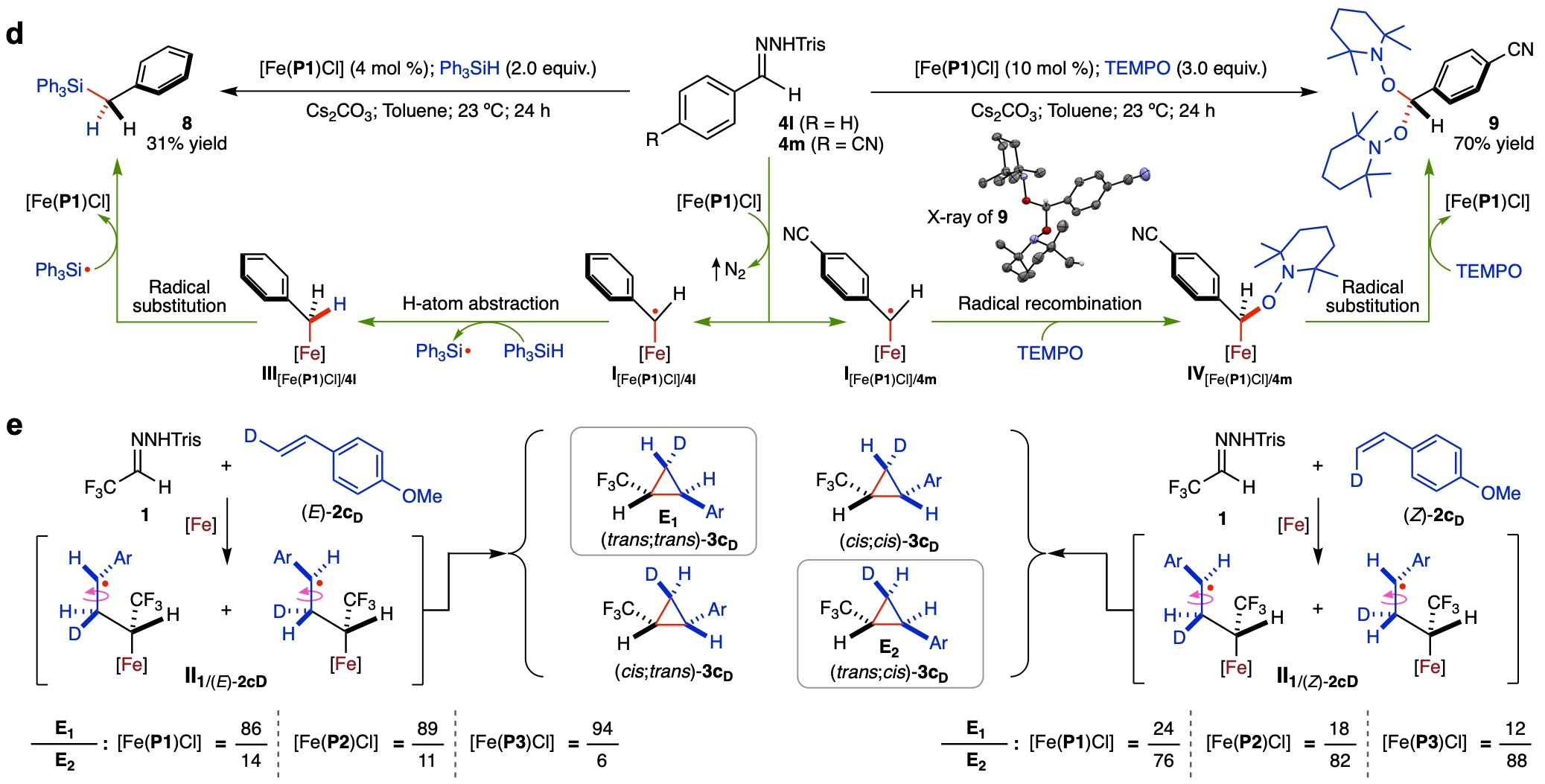
Fig. 3. Experimental studies on the catalytic mechanism of olefin cyclopropanation by the Fe(III)-based metalloradical system
In summary, we have explicitly demonstrated the operation of Fe(III)-based MRC in the context of its application for asymmetric olefinm cyclopropanation. Supported by the D2-symmetric chiral amidoporphyrin 3,5-DitBu-QingPhyrin as the optimal ligand, the Fe(III)-based metalloradical system, which operates under mild conditions in a one-pot fashion with in situ-generated α-trifluoromethyldiazomethane, is highly effective for radical cyclopropanation of a wide range of alkenes, affording trifluoromethyl-substituted cyclopropanes in high yields with both high diastereoselectivity and enantioselectivity. Additionally, we have shown that the Fe(III)-based metalloradical system is broadly effective in activating different classes of diazo compounds for asymmetric radical cyclopropanation, offering a potentially general and practically sustainable approach for stereoselective synthesis of valuable three-membered carbocycles. Our systematic experimental studies, in combination with detailed DFT computations, provide multiple lines of convincing evidence supporting the genuine action of Fe(III)-based metalloradical catalysis and shed light on the underlying stepwise radical mechanism. Specifically, we have established that the five-coordinate Fe(III) complexes of porphyrins ([Fe(Por)X]), a class of stable 15e-metalloradical complexes, can function as potent metalloradical catalysts that have the ability to homolytically activate diazo compounds and generate α-Fe(IV)-alkyl and γ-Fe(IV)-alkyl radicals as catalytic intermediates. Beyond cyclopropanation, our ongoing research aims to explore the reactivity of α-Fe(IV)-alkyl radicals, in H-atom and X-atom abstraction reactions. We believe that the outcomes presented here will lay a solid foundation for Fe(III)-based metalloradical catalysis and pave the way for the development of novel stereoselective radical transformations. This work represents an important step towards advancing the field of metalloradical catalysis and holds promise for the design of efficient radical methods for stereoselective organic synthesis.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in