Explore the Research
Utility of large subunit for environmental sequencing of arbuscular mycorrhizal fungi: a new reference database and pipeline
Click on the article title to read more.
I was asked recently: “How did you learn bioinformatics?”. The answer is not one class or one turning point. Instead, I ‘learned bioinformatics’, or rather, started learning bioinformatics, with a lot of effort over the past 4 years. It all started in the lab office at the University of Kansas at the start of my PhD. A post-doc taught us all she knew. We met once a week to code and mess up together. Half of us, me included, were new to Linux and command line, using a computing cluster, and thinking about massive sequencing datasets. It was a steep learning curve. We asked a lot of ‘stupid’ questions, and did a lot of online searching for solutions. But, the goal of wrangling sequence files to answer our questions kept us going (Fig 1.)
This work was centered around bioinformatics to clean and prepare arbuscular mycorrhizal fungi (AMF) sequences for analysis. But, I was also working with other types of fungi and the first in my lab to work with oomycetes (an important group of plant pathogens). I reached out to another professor working with fungi. He shared his entire script for fungal analyses without a second thought. I also happened to have a master bioinformatics post doc behind me in my office space. Once I got through the pipeline with help from everyone above and more, I came upon implementing FUNGuild to assign likely guild/ functional assignments to fungi. Good thing another post-doc who had worked with this database, was across from me in the office and helped me submit my first database match. I then adapted the fungal pipeline for oomycetes, probably messaging the post-doc who made the primers I was using way too much. Then, came the phylogenies. Here, I leaned on another PhD student, my advisor, and researchers I’d never met on the RAxML google group. Also, can’t forget the amazing people at the university cluster that dealt with my never-ending barrage of questions and issues, whether about the cluster or just general command line. What is a parallel job? How do I send one on multiple cores, and on which partition?
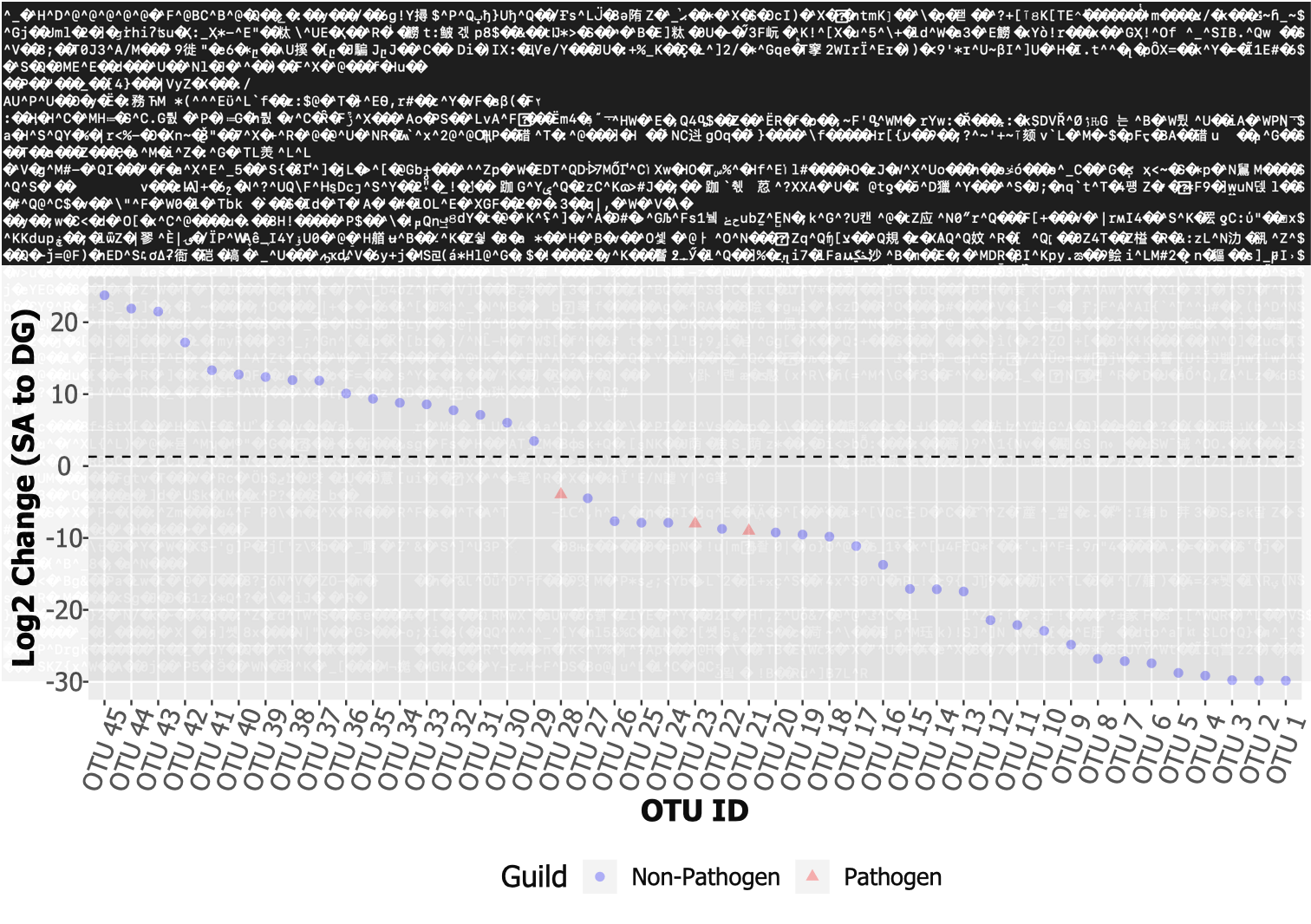
Figure 1. From the mess of a .fastq file (top; from the sequencer) to actual inference about the samples (bottom). Here, we are determining which taxa are differentially over- or underrepresented in the species "SA" compared to "DG", and how this differs between guilds.
I knew I had made tremendous progress from the days sitting in the lab office with a bunch of friends and colleagues trying to get each command to run without error, but I wanted to take it up a notch. So, I contacted a new professor in our department, since I knew she was planning to teach a microbiome class involving bioinformatics. I thought, if I can learn this enough to teach it, then I’ll really know it. It was an incredible experience, working together to plan what would be included in the class, creating the class lectures and hands on activities, condensing what we knew into presentations, and bringing together all the resources and people we could think of to students (find our online repository for the course here!).
Working with arbuscular mycorrhizal fungi is a bit challenging, since many of them have not been identified. This makes working with environmental samples difficult; how can we determine what is an arbuscular mycorrhizal fungus if it’s likely the majority of our taxa have never been seen before, and can’t be found in a database? Early on in my PhD, I used a custom Python pipeline built by another post-doc and then built phylogenetic trees to place these fungi in their phylum, identifying even unknown taxa. But, scrolling through 2000-tip trees on a computer screen, no matter how big, is tedious and error prone. What if we improved our lab database, updated our pipeline to be used in a common bioinformatics program and made the phylogenetic placement automated? My goal here was to make a pipeline that would be usable by me three years ago. And, with a super strong team of collaborators, that’s what we did (see paper here).
To answer the original question. I learned bioinformatics with help from a lot of amazing people and resources, by trial and error, teaching, and experience. Despite my progress, I’m daily reminded of what’s yet to learn. Parallelizing commands within R on the cluster? Nested for loops using sequences and metadata? Pruning trees across a million plots in one go? I think this is part of why I love science: the incessant reminder of all that you don’t know. This reminder means you’re doing it right. Pushing boundaries isn’t comfortable, but it’s how we learn.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in