Linking genome architecture and antigenic variation
Published in Microbiology
The paper can be found here: doi.org/10.1038/s41586-018-0619-8
My group studies how changes in chromatin allow parasites to evade the host immune response via antigenic variation (http://siegel-lab.de). To address this question, we use Trypanosoma brucei, a deadly parasite that causes sleeping sickness in humans. Already when I was a postdoc, I was thinking about how to best study genome architecture in this parasite. I had experience in different deep sequencing-based approaches, yet it was clear to me that establishing a pipeline to determine genome-wide DNA-DNA contact frequency using Hi-C technology presented a new level of complexity.
Two incidents encouraged me to finally investigate the genome architecture. First, I was able to convince Laura Müller, a student at the University of Würzburg, to establish Hi-C as her master project in my lab (I am sure I was only able to convince her because I had severely underestimated the effort it would take us to finish the project). Second, Job Dekker, who had ‘invented’ Hi-C and similar assays, offered to help us establish the technology in T. brucei.
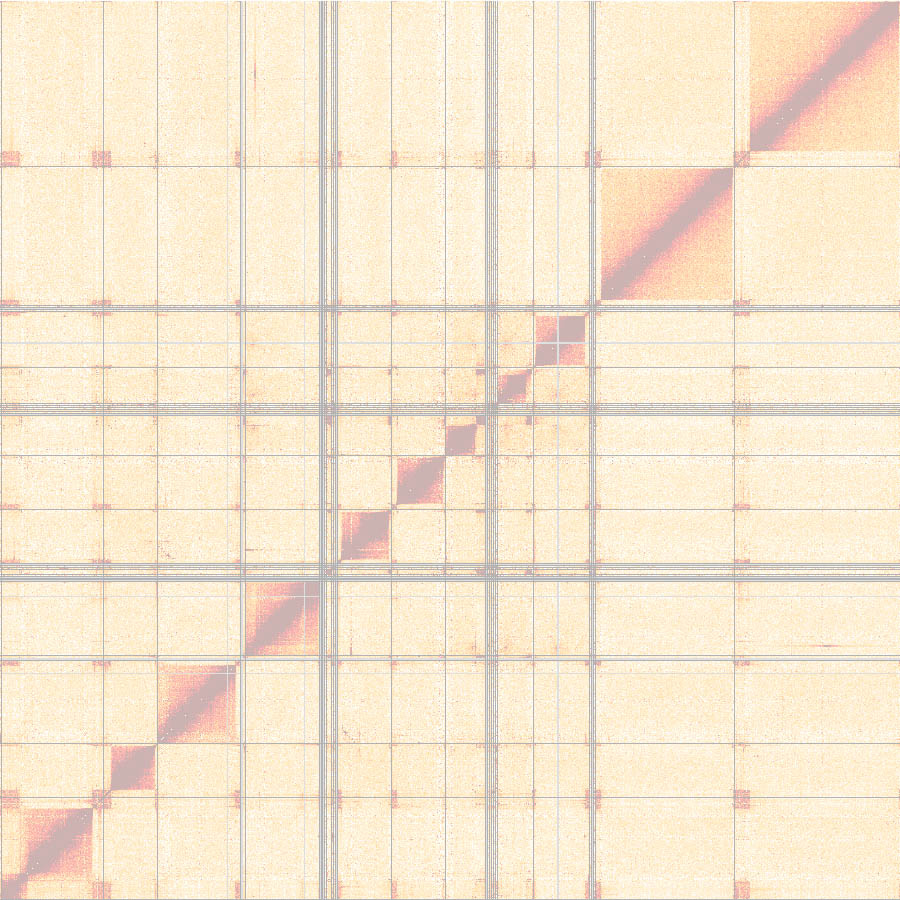
Our first Hi-C datasets looked promising, but we quickly realized that the genome we were using was inadequate – it was missing most antigen-coding genes. During discussions with members of the Dekker lab, it was Noam Kaplan (now a professor at the Technion Israel Institute of Technology in Haifa) who convinced us to generate a de novo assembly and to use the Hi-C data for the scaffolding. While we liked the idea very much, it meant that we had to take a major step back. Instead of studying the 3D organization, we were finding ourselves piecing together the linear genome.
Clearly, the project was becoming too complex for one master student. At this point we were fortunate that Raúl Cosentino joined our lab and that Laura continued her work as a PhD student. Now we had two people working full time on the project. Raúl was putting together the linear genome and Laura was looking at the 3D architecture. The assembly of the genome was quite exciting. For the first time we were able to place the sites of antigen expression into the genomic context.
The problem with having one person working on the genome assembly and one person using the genome for downstream analyses was that every time Raúl improved the genome, Laura had to start her analyses again. I don’t think we would have ever finished the project without the help of Konrad Förstner, now joint professor at ZB MED and the TH Köln - University of Applied Sciences. Konrad spent endless hours with Laura figuring out how to best analyze our Hi-C data and, equally important, to script every step of our analyses so that it could be quickly rerun and reproduced. In addition to the constant changes, it was the partially phased nature of our genome assembly that presented a challenge: the core, coding for housekeeping genes, is diploid, and the subtelomeres, containing long arrays of genes coding for antigens, are haploid.
Laura and Konrad in the process of setting up pipelines to analyze our Hi-C data.
Laura’s PhD project was becoming more and more a team effort and we received a lot of support from researchers within the T. brucei community and beyond. It was only thanks to their help that we were able to conclude our story and we are very excited about our findings that histone variants link genome architecture and antigenic variation in African trypanosomes. Nevertheless, it is clear that many questions regarding antigenic variation remain to be addressed. We are, for example, very intrigued by the strong bias for the activation of particular antigen following the switch in antigen expression or, why deletion of one histone variant has a strong impact on genome architecture while the deletion of another histone variant that shows a similar genome-wide distribution has none.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Nice post on a fantastic paper! Out of curiosity, I noticed that you didn't use the word epigenetics in the paper. Would you consider this to be epigenetics or something different?
We think that the preference for some antigens over others may indeed be mediated by some epigenetic factors.