Meningococci exploit plasma membrane wetting to trap host receptors and initiate infection
Published in Chemistry, Microbiology, and Cell & Molecular Biology
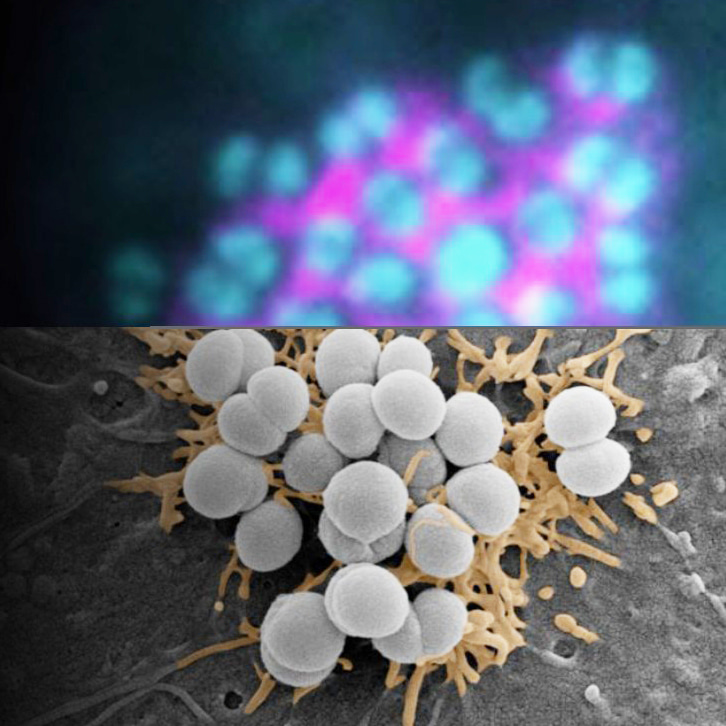
Neisseria meningitidis (meningococcus), the causative agent of meningococcal septicemia and meningitis, faces a critical challenge: how to rapidly and efficiently adhere to host endothelial cells despite the shear stress of blood flow and the low probability of ligand-receptor encounters, before being eliminated by the innate Immunity of the host. Unlike soluble ligands, bacterial adhesins and ligands for specific host receptors are anchored to the bacterial surface, drastically reducing their effective concentration and limiting their ability to engage their receptors. To overcome this constraint, N. meningitidis exploits a biophysical mechanism. Bacterial appendages, the type IV pili (T4P), induce the formation of tubular membrane structures (TMS) on host cells through a signaling-independent, wetting-driven process. These TMS non-specifically concentrate plasma membrane proteins—including key adhesion and signaling receptors—thereby facilitating bacterial colonization. This mechanism not only explains the remarkable efficiency of meningococcal adhesion but also introduces a new paradigm for pathogen-host interactions, where physical forces shape the early stages of infection.
A Paradigm Shift in Pathogen Adhesion
The study challenges the conventional view that bacterial adhesion and subsequent signaling in host cells relies solely on high-affinity ligand-receptor interactions. Instead, it shows that membrane wetting, a physical process driven by the interaction between T4P and the host plasma membrane, is the initial step in receptor recruitment. Unlike previously described adhesion mechanisms, this process operates in the absence of host cell signaling, ATP, or cytoskeletal remodeling, as demonstrated in fixed cells and purified membrane sheets. By extending the contact interface and locally enriching receptors, TMS increase the likelihood of productive interactions between bacterial ligands (e.g., PilV) and host receptors (e.g., CD147/β2-adrenergic receptor complexes). This mechanism may be conserved across bacterial species that use pili or fimbriae for adhesion, such as Pseudomonas aeruginosa, uropathogenic E. coli, and Streptococcus pneumoniae.
Implications for Infectious Disease
The findings provide a molecular explanation for the exceptionally low infectious dose of N. meningitidis (as few as five diplococci can initiate septicemia in murine models). By concentrating receptors in TMS, the bacterium ensures stable adhesion even under shear stress, a critical step for vascular colonization and disease progression. This work also highlights the PilC1 adhesin as the initiator of membrane tubulation, while PilV reinforces adhesion by engaging CD147/β2AR complexes. Understanding this process could inform the development of novel anti-adhesive therapies targeting T4P or their host receptors, potentially disrupting the early stages of infection. However, the unknown PilC1 receptor and the redundancy of adhesion pathways in some pathogens remain obstacles.
Key Experimental Findings
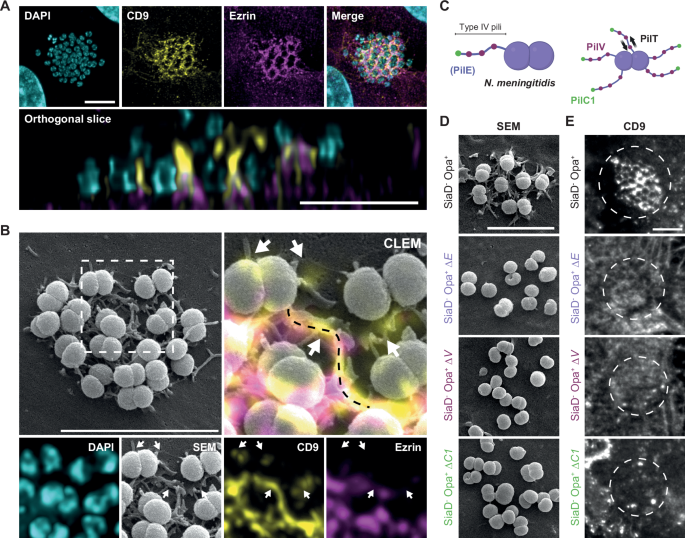
- Using correlative light and electron microscopy (CLEM), we showed show that TMS formed after bacterial contact are enriched in plasma membrane proteins such as CD9, CD81, CD151, CD147 (the receptor essential for stable adhesion) and the β2-adenergic receptor (β2AR, which activates meningococcal-dependent intracellular signaling pathways).
- Strikingly, TMS assembly persists even in paraformaldehyde-fixed cells—where all enzymatic activity is abolished, and in ATP-depleted cells or those treated with actin inhibitors (cytochalasin D, latrunculin B). This robustness was further confirmed using purified plasma membrane sheets, which lack cytoskeletal attachments entirely. Together, these data unequivocally decouple tubular structure formation from host cell metabolism or structural dynamics.
- FRAP experiments further assessed that the diffusion of some membrane proteins (e.g., CD9, β2AR, CD4) is significantly reduced within TMS, leading to their local accumulation.
- PilC1, the adhesin located at the pilus tip, is essential for initiating membrane tubulation. Indeed, mutants lacking PilC1 (ΔC1) failed to induce TMS, even when PilV is expressed. PilV, distributed along the pilus, interacts with CD147/β2AR complexes to stabilize adhesion and trigger downstream signaling.
- While TMS formation is signaling-independent, their stabilization and enlargement depend on actin polymerization and ezrin recruitment. Inhibiting ezrin phosphorylation (NSC668394) or actin polymerization (cytochalasin D, latrunculin B) reduces the size and stability of TMS, highlighting a two-step process: the PilC1-dependent, signaling-independent physical tubulation, which precedes the PilV/β2AR-dependent, signaling-dependent cytoskeletal reinforcement.
- Tubular srtuctures indiscriminately trap membrane proteins, including other G protein-coupled receptors (e.g., CXCR4, CCR5) and single-pass transmembrane proteins (e.g., CD44, CD4), but exclude others (e.g., transferrin receptor, TLR 2/4); membrane curvature and lipid composition (e.g., cholesterol, sphingolipids) might qualitatively influence protein recruitment.
We propose a three-step model to describe how meningococcus recruits and activate its cognate receptors:
- Initiation: PilC1 binds to endothelial cell membranes (via a host receptor that is still unknown, probably present in numerous copies, such as a sugar), triggering membrane wetting and TMS formation.
- Receptor clustering: Tubular structures concentrate CD147/β2AR complexes near bacterial ligands, facilitating PilV engagement.
- Stabilization: T4P retraction and blood flow apply mechanical forces to β2AR, activating signaling pathways that recruit ezrin and actin, reinforcing tubular structures and promoting bacterial colony growth.
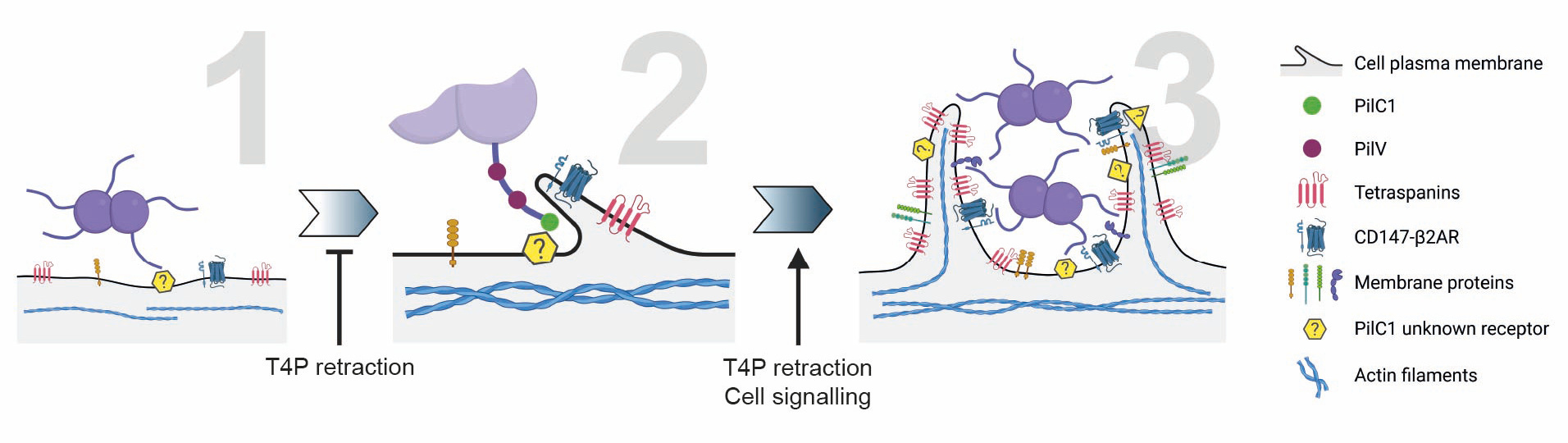
This model integrates biophysical and biochemical cues, emphasizing the synergy between physical forces and molecular interactions in pathogen adhesion.
The discovery of wetting-driven receptor recruitment raises questions about its conservation across pathogens and potential roles in non-infectious processes (e.g., cell-cell adhesion, immune synapse formation). Future studies could explore whether other piliated bacteria (e.g., P. aeruginosa, E. coli) exploit similar mechanisms.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in