Live-cell imaging of chromatin loops in an unmodified genome
Published in Bioengineering & Biotechnology

| The paper⇒ Clow et al CRISPR-mediated multiplexed live cell imaging of nonrepetitive genomic loci with one guide RNA per locus https://www.nature.com/articles/s41467-022-29343-z |
Introduction - the 3D genome
Within each of our 37.2 trillion cells, at the center, is the command center called the nucleus which is around 10 micrometer (hundred-thousandth of a meter) in diameter. Our genetic blueprint called the genome, a collection of genes, is written on strands of DNA totaling three meters long. The length of the genome relative to the small nuclear space means that it has to be folded extremely tightly. Genes are like computer files, the cell sometimes opens some of these files so that the instructions can be read to make appropriate proteins. There is a master lock called “promoter” positioned right next to the gene on the DNA strands and several other locks called “enhancers” positioned far away. Enhancers and promoters together determine whether the gene file is open. Proteins called transcription factors bind to enhancers and promoters act as keys to open the genes. Enhancers are usually located far away from the promoter in the DNA strand and need to be relocated close to the promoter in the 3D space to turn on genes. It is thus obvious that the folding of the genome is important to determine what genes (“files”) are read at any given time and ultimately the function of the cell. Since life needs to respond to constant changes, genome folding is not fixed, but is instead constantly changing over time.
Sequencing technologies for 3D genome
During the past decades, scientists have developed sequencing methods to determine the DNA sequence of the genome in “linear order”, reading from the first letter to the last. In the 2000s, Sequencing methods have been cleverly tweaked to identify sequences that are close in 3D instead. It involves fixing cells with chemicals (thus freezing the cells in time), chopping up the genome in place, and rejoining DNA fragments close to each other in the 3D space by a technique called “proximity ligation”. Hi-C, ChIA-PET, HiChIP, etc., are different methods for sequencing the 3D genome. These methods take in millions of cells and mix all their genomes together. However, individual cells are not the same, therefore these methods give “average” interaction frequencies between pairs of loci. Since the time is frozen, they cannot track the dynamic of genome folding within the otherwise living and constantly changing cells. Furthermore, because data are only generated for genomic locations that are close in 3D space, but not when loci are far apart, it is hard to translate interaction frequencies into actual 3D coordinates.
Imaging technologies for 3D genome
Another way to look at genome structure inside the nucleus is by imaging. One of the most routinely used techniques in cell and molecular biology called DNA fluorescent in-situ hybridization, in short DNA-FISH, uses fluorescent dye-coupled DNA probes that can anneal with the genome at the target locations. DNA-FISH again starts with fixed cells and requires denaturing genomic DNA, in order to let dye-attached DNA probes go into the genome and anneal to matching sequences. This method can be used to tell the actual locations of genomic loci in 3D coordinates. However, since cells are frozen in time, it cannot track the dynamic motion of DNA over time.
Live-cell imaging is thus the most direct way to observe the motion of DNA and the changes of genome folding over time, in the context of living cells. It was first demonstrated by Dr. Carmen Robinett in Andrew Belmont lab in 1996 by engineering the LacO system. (Fun fact: Dr. Robinett is currently at Jackson Laboratory. I read and cited her paper, and have worked with her on other unrelated projects. One day I talked to her about our imaging work and told her about the pioneering work using the LacO system, and there I found out that the pioneer of live-cell genome imaging is sitting right next to me!). In this pioneering paper, LacI protein is fused to GFP, while the target genomic location is inserted with 256 copies of LacI’s binding sites (LacO). Many molecules (up to 256) of LacI-GFP binds to the target location and form a fluorescent spot which under live-cell microscope moves with the target genomic locus. Following this work, others (e.g., Alexander et al) have engineered imaging systems based on GAL4, CuO, TetR, etc., and with these orthogonal DNA binding proteins fused with different fluorescent proteins, multiple loci imaging was achieved. These methods enable the first visualization of dynamic changes in 3D genome. However, large tandem arrays containing hundreds of binding sites need to be inserted into each target, which is tedious and may disrupt the normal folding and functions of the target loci.
ZFP, TALE and CRISPR for genome imaging
Labeling native, unmodified genomic loci would thus be ideal. This is enabled by the discovery and successful re-engineering of zinc finger proteins (ZFP), Transcription Activator-Like Effector (TALE) and more recently the CRISPR systems. ZFP and TALE are proteins that bind to genome by recognizing specific sequences. They do so through modular protein parts each recognizing a DNA triplet (for ZFP) or one nucleotide (for TALE). Protein modules can be swapped in or out like LEGO blocks to construct new ZFP and TALE for recognizing particular sequences. Thus, ZFP and TALE can be tailored to bind to specific locations in the genome with a defined sequence and can bring with them fluorescent proteins by protein fusion (e.g., Lindhout et al, Miyanari et al). With these ZFP- and TALE- fluorescent protein fusions, researchers can label unmodified genomes. However, every new target requires designing and cloning of new ZFP and TALE proteins, therefore limiting the throughput of the methods. Another limitation is that since one molecule of fluorescent protein is brought to the target location, these methods were restricted to highly repetitive sequences (such as Telomeric or centromeric repeats) where hundreds of copies of target sequences allow concentration of many molecules of fluorescent proteins to generate a bright spot.
In 2013, CRISPR shocked the world when scientists demonstrated that a nuclease (e.g., Cas9) can bind to and cut DNA in the genome using short RNA sequence as guide RNA. In addition to a short motif on the target DNA called protoadjacent motif (PAM), the Cas9 enzyme finds it target complementary to the "spacer" sequence on the guide RNA. It means that we can easily address the genome and bring molecules to specific places of the genome in living cells (My favorite analogy is comparing CRISPR to “find-and-replace” function in Word). In addition to cutting/editing DNA, CRISPR has been engineered to specifically turn on and off genes, to change epigenetic states of genomic regions (like tagging different files with different labels in Finder, so that the cell remembers and knows how to interpret specific "files” (genes) in the genome), change how RNA is processed, even to detect or destroy SARS-CoV-2 viruses (the virus that caused the COVID-19 pandemic). It is not surprising that CRISPR provides innovation in 3D genome biology, too. In 2013, Chen et al demonstrated the fusion of nuclease-deficient Cas9 (dCas9, a mutant version that binds but not cut DNA) to GFP allows RNA-guided live-cell labeling of genomic loci. Again, because only one GFP is brought to a target site, the technique is well-suited for labeling repetitive sequences. Given the easily programmable feature of CRISPR system, however, one can introduce to cells 36 pieces of guide RNA to direct 36 molecules of dCas9-GFP to bind to nearby sites, to produce a detectable spot, allowing even non-repetitive sequences to be labeled. However, delivering 36 pieces of guide RNA can still be challenging and labeling with one color does not give very much information about genome folding. Since then, the holy grail of the field is to reduce guide RNA requirement as well as to allow multiple locations of the genome to be visualized simultaneously.
One way to reduce guide RNA requirement is to recruit many more fluorescent protein molecules to the dCas9 complex. In 2014, Tanenbaum et al reported an approach called “SunTag” in which dCas9 is fused to 24 repeats of short peptide antigens from GCN4 protein, which is then bound by anti-GCN4 antibodies that are fused to GFP, essentially concentrating fluorescent signal at the target site. However, this system does not allow multicolor labeling of multiple locations which instead would require orthogonal Cas proteins as well as orthogonal antigen-antibody pairs.
Casilio - the molecular operating system for the genome
Most of the engineered CRISPR tools were focused on one specific functionality in 2014 when I joined the Jackson Laboratory as a JAX Scholar in the lab of Dr. Haoyi Wang. (Haoyi and I met at Rudolf Jaenisch lab where he was a postdoc and I was a graduate student and we later both moved to the Jackson Laboratory). We thought we could build an “operating system” (OS) for the genome, very much like the OS on our smart phone or our computers, on which “apps” can be developed and installed, each providing a specific functionality. Most importantly, apps can run simultaneously (“Multitasking”). In cells, it means that with such a molecular OS, one can activate a set of genes, repress another set, modify histones at yet another set of genes, etc. This would be useful for modulating regulatory networks in complex developmental and pathological processes. We came up with a modular design we named “Casilio”, combining “Cas” from (d)Cas9, “ilio” from Pumilio, which is a family of RNA binding proteins conserved across eukaryotes. In Casilio, effector domains are not directly fused to dCas9, but are rather fused to Pumilio/FBF (PUF) RNA binding domains (RBD) borrowed from Pumilio proteins. PUF-RBD are like TALE for RNA. Like TALE, PUF-RBD contains peptide subunits each recognizing an RNA base. One can change two residues in each subunit to program it to recognize one of the four RNA bases. An 8-subunit PUF-RBD, as in the case of human Pumilio proteins, can be programmed to recognize virtually any of the 48 = 65,536 octamers. Every new Casilio effector can be assigned to (i.e., fused to) a PUF-RBD variant. To link a PUF-effector to the dCas9 complex, the guide RNA is appended with the corresponding octamer sequence. A guide RNA with Pumilio Binding Site (gRNA-PBS) pairs the CRISPR genomic address (the 5’ spacer portion) with effector selector (the 3’ appended Pumilio Binding Sites) such that a specific effector can be recruited to each target by dCas9 via the guide RNA. We showed that we could activate and repress two different genes simultaneously (multitasking/multiplexing). By placing multiple copies of pumilio binding sites on the guide RNA, we were able to recruit different numbers of effector molecules to the target (multimerization). We also provided proof-of-principle data for Casilio-mediated genomic imaging. Using the multimerization and multiplexing features, Casilio can increase signal strength as well as allow dual-color labeling of telomeric and centromeric repeats in the same cell.
 In Casilio, gRNA is appended with one or more binding sites for Pumilio/FBF (PUF) RNA binding domain-tethered effectors. Each PUF domain contains 8 peptide subunits each programmable to recognize one of the four RNA bases. A collection of PUF-effectors can be created by fusing different effectors to PUF programmed to recognize different 8-mer PUF binding sequence (PBS). Effectors are recruited to dCas9/gRNA-PBS complex according to the fused PUF domain. (b) Casilio architecture allows multiple molecules of the same or different effectors (E1, E2, …, etc.) to be recruited to a target (multimerization or complex formation) as well as different set of effectors to be recruited at different targets in the same cell (multiplexing). This system is expandable to potentially over 65,000 possible effectors.")
In 2016, we published the Casilio paper, and in the same year, Fu et al and Ma et al published guide RNA scaffold-based imaging systems based on MS2, PP7 and boxB. At this point, although the three papers showed that signal amplification as well as dual- or multi-color labeling can be achieved by scaffolded guide RNA, imaging experiments were only conducted on repetitive sequences. In the following years, different labs used similar RNA scaffold strategies and attempted to reduce the guide RNA requirement. In 2017, Qin et al used the MS2 scaffold system to achieve four-guide RNA labeling of non-repetitive regions. In 2018, Mass et al used MS2 and PP7 systems and demonstrated two-guide RNA labeling of non-repetitive regions. Although MS2 and PP7 are useful scaffolds, Ma et al found that, as the copy number of MS2 on guide RNA increases to 14, its expression is significantly reduced.
Finally, about this paper
the holy grail of the field is to reduce guide RNA requirement as well as to allow multiple locations of the genome to be visualized simultaneously.
When Dr. Patricia Clow joined my lab in 2017, we thought we could give Casilio a try at nonrepetitive labeling since we knew in our first Casilio paper in 2016 that adding up to 47 copies of pumilio binding sites to guide RNA did not significantly affect dCas9 binding activity. We set out to try 15 copies of pumilio binding sites on guide RNA and started on labeling MUC4, the first gene labeled by CRISPR-based imaging method. To our surprise, we were able to get fluorescent spots with one guide RNA. At first I did not believe the results so we performed DNA-FISH, and encouragingly yes, those spots are specific. To further confirm our observation, we conducted several other control experiments, including no-dCas9 control, cells with different ploidy, as well as competitor experiments.

| A side story - In 2018, the Verhaak lab was looking for ways to label extrachromosomoal DNA (ecDNA) in live cancer cells to study how they segregate during cell divisions. These ecDNAs are circular DNA that are outside of the chromosomes. EcDNAs carry oncogenes and can multiply to up to hundreds of copies of ecDNAs in a single cell, and as a result confer growth advantages or drug resistance. Due to the mechanisms of the formation of ecDNA via circularization and joining of different genomic fragments, they present unique junctional sequences that are not found in the genome, representing unique opportunities to label them. However, previous CRISPR imaging systems require multiple sites to achieve enough signal. Given the success in our MUC4 one-guide labeling, we proposed to the Verhaak lab that Casilio might be able to label these ecDNAs with one guide RNA. By designing Casilio guide RNAs to label ecDNA junctional sequences, the “ecTag” method based on Casilio was born. Live-cell imaging of ecDNA with ecTag revealed their uneven segregation patterns during cell division as well as the dynamic hub-forming behavior. This work was published recently at Cancer Discovery. |
In the past, the high guide RNA requirement for non-repetitive sequence labeling as well as the lack of a multi-color system for nonrepetitive sequence imaging limited the adoption of CRISPR imaging systems for studying dynamic chromatin interaction in live cells. Our positive result from MUC4 motivated us to further develop the system to label more loci simultaneously and start looking at chromatin interactions. We selected a chromatin interaction between MASP1 and BCL6 genes from ARPE-19 ChIA-PET dataset on the ENCODE database. We labeled one anchor with Clover and another with iRFP670. And voila! It was very exciting to observe two native genomic loci move with respect to each other using Casilio the first time!
Next, we asked whether we could use this to study the functions of architectural proteins on chromatin interactions. Natsume et al developed an inducible protein degradation system called auxin inducible degron (AID). AID is a protein domain that when fused to a host protein can trigger rapid degradation of the protein in response to auxin. Coding sequence of AID was knocked-in in frame and downstream of both alleles of endogenous RAD21 (a subunit of cohesin) in HCT-116 cells allowing these cells to be depleted of RAD21 in as fast as six hours with auxin added to the media. This HCT-116 RAD21-mAID cell line was used by Rao et al as a model to study chromatin loop in the absence of RAD21. Rao et al reported that cohesin loops including many connecting super-enhancers and their target genes were depleted upon RAD21 degradation. Interestingly, another class of “RAD21-independent” loops behave in an opposite way, that is, their interaction increases upon RAD21 depletion. We therefore selected one loop from each class and applied Casilio imaging to track chromatin interactions in the presence or absence of RAD21. As expected, the distance of the IER5L promoter and its super-enhancer increases in the absence of RAD21 consistent with sequencing showing RAD21-dependency. On the other hand, the distance between interaction anchors of a RAD21-independent loop decreases, recapitulating the increase in contact frequency in sequencing data in the absence of RAD21.
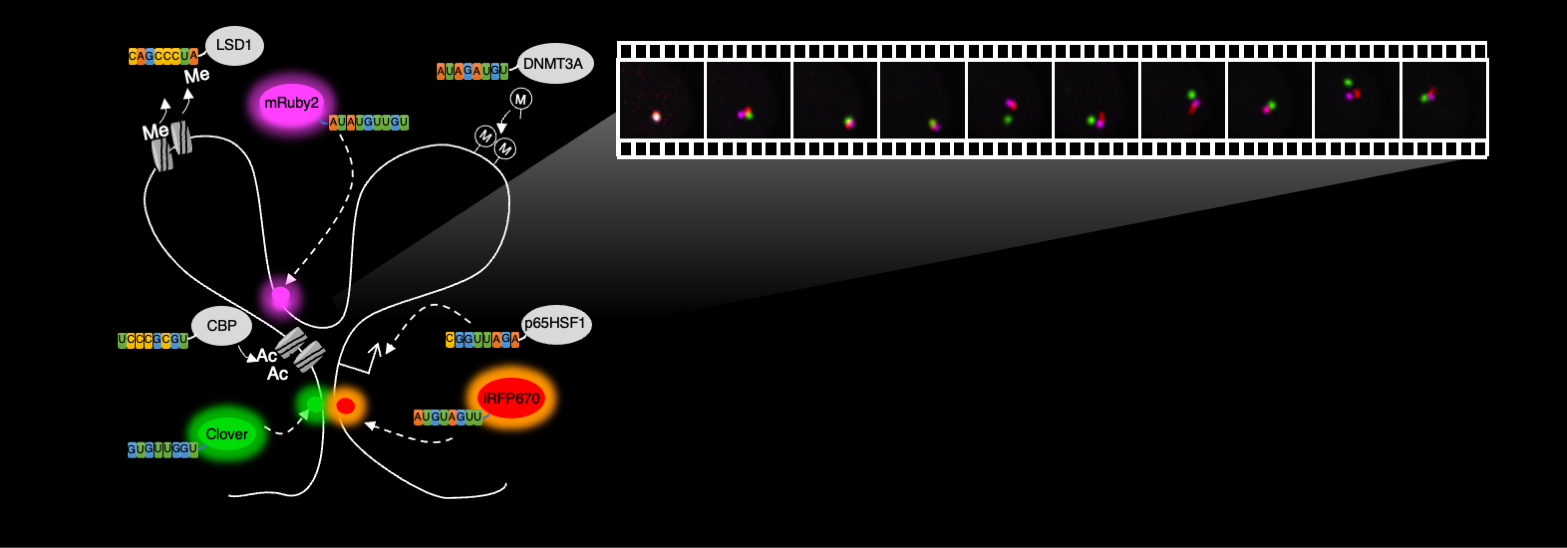
Next, we wanted to test the idea of placing more probes between chromatin interaction anchors with the ultimate goal of deploying enough probes along the chromatin loop to allow for the visualization of loop extrusion processes as it happens in live cells. We added a third color to the Casilio “palette”, mRuby2 and placed Clover, iRFP670 and mRuby2 at the IER5L promoter, mid-point, and super-enhancer, respectively. This experiment revealed dynamic motions of several reference points along a promoter-enhancer loop, suggesting that loops are more dynamic than what was generally assumed.
| A software for CRISPR designs - An important aspect of genome imaging is the design of targeting sequences. In a separate work, described in a BioRxiv preprint, we developed a software package JACKIE for efficiently enumerating single- or multi-copy CRISPR binding sites and their off-target predictions over the genome, allowing the generation of browser tracks that can be loaded onto genome browsers alongside other tracks for convenient design of imaging experiments. We provide single-copy guide RNA databases for use with Casilio imaging experiment, as well as multicopy CRISPR site clusters for use with other imaging methods requiring clustered binding sites. Visit https://cheng.bio/JACKIE for more details and links to UCSC Genome Browser sessions containing guide RNA tracks. |
In the future..
In this paper, we only demonstrated three point imaging for chromatin loop, we are therefore still far from tracing the entire loop structure to study processes like loop extrusion. In the future, we will have to develop more colors for Casilio and deploy more probes along chromatin loops. Given the expandability and multitasking capabilities of Casilio, we envision that future experiments can employ Casilio epigenetic editing and imaging modules concurrently, so that epigenetic changes (e.g., DNA methylation/demethylation) can be induced at cis-regulatory elements such as enhancers or CTCF sites, while simultaneous imaging can read out resultant dynamic changes to chromatin structure “on the fly”. We hope that Casilio will be used to uncover many new insights for the 4D nucleome.
, acetylating histone (CBP), methylating DNA (DNMT3A), activating (p65HSF1) different targets, and at the same time monitoring multiple locations with different fluorescent proteins in live cells. dCas9/gRNA-PBS not drawn for simplicity.")
The potential for Casilio to achieve multimodal operations simultaneously, for example, demethylating histone (using LSD1 module), acetylating histone (CBP), methylating DNA (DNMT3A), activating (p65HSF1) different targets, and at the same time monitoring multiple locations with different fluorescent proteins in live cells. dCas9/gRNA-PBS not drawn for simplicity.
| The paper⇒ Clow et al CRISPR-mediated multiplexed live cell imaging of nonrepetitive genomic loci with one guide RNA per locus https://www.nature.com/articles/s41467-022-29343-z |
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in