Long-chain hydrocarbons from carbon dioxide using polarized nickel electrocatalysts
Published in Chemistry

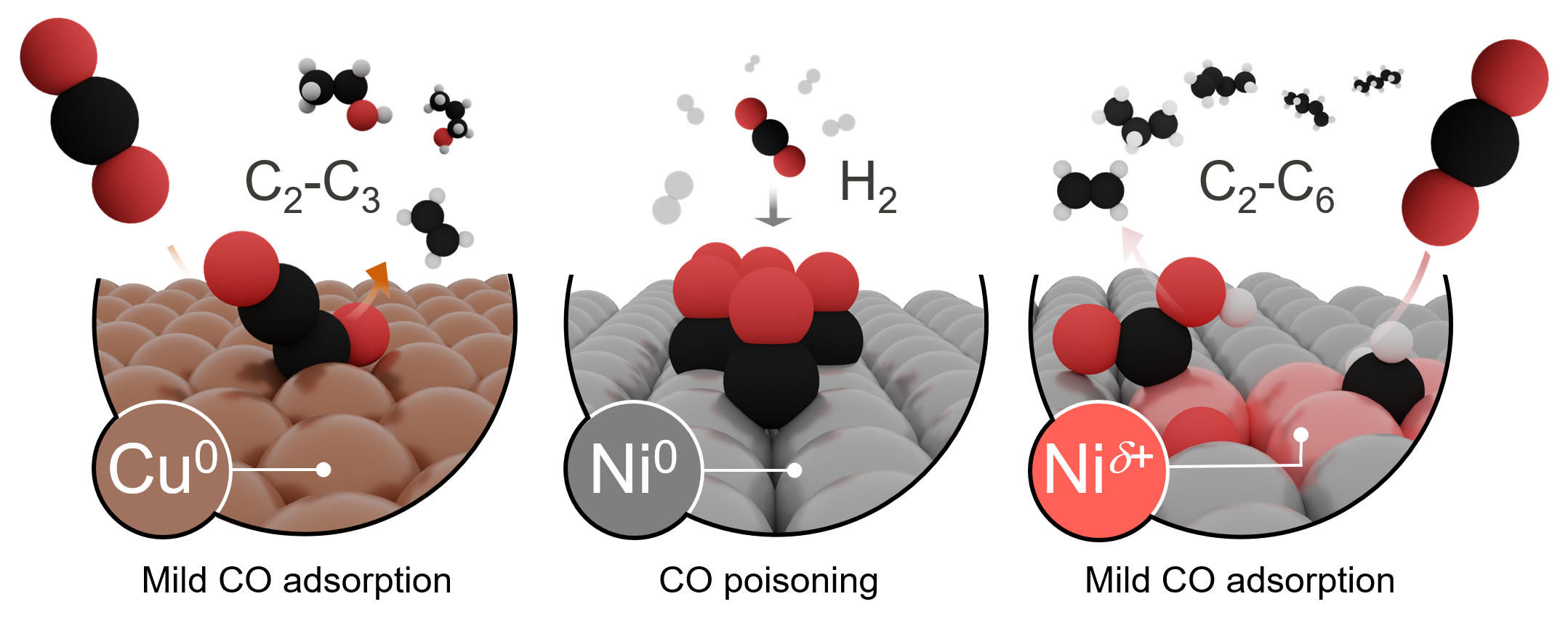
Although the electrochemical carbon dioxide reduction reaction (eCO2RR) using renewable electricity is seen as a way to close the carbon cycle, the reality is that after 40 years of research only lab-scale setups using copper catalysts and producing mainly simple molecules such as ethylene have been developed.1,2 (Fig. 1, left). The potential of nickel metal as an eCO2RR catalyst has been mostly ignored due to its known propensity to strong CO poisoning and deactivation (Fig. 1, middle) already hinted in the seminal screening of metals.3 Nonetheless, the sparse examples of reports dealing with nickel-containing catalysts do have something in common: formation of tiny amounts of multicarbon products.4-6 This sparked our curiosity and triggered our interest to explore various families of nickel compounds.
Ready for a challenging product quantification, we performed our first tests on metallic nickel that unexpectedly produced several C2-C5 hydrocarbons, albeit at low Faradaic efficiencies (FEC2+ ~ 0.3%). It was clear there were nickel active sites not poisoned by CO, but, how to make them more abundant? Testing a NiO-modified nickel disk was good decision, for which we obtained a still small but certainly higher FEC2+ ~ 0.8%. As the excitement dawned, other oxygen-containing nickel compounds entered the testing program like nickel phosphate, carbonate, borate, and bicarbonate. We coined them as inorganic nickel oxygenate-derived electrocatalysts, or INOs. Et voilà. The performance of all INOs catalysts was strikingly high. For example, the nickel phosphate-derived catalyst produced 29 products, reaching ca. 15% FE toward C2+ products at –1.2 V vs RHE in a flow cell using 0.1 M KHCO3, with a large fraction (6.5 %) corresponding to C3-C6 hydrocarbons. As long hydrocarbons readily brings the Fischer-Tropsch process to mind, we analyzed their distribution to learn it indeed followed the Anderson-Schulz-Flory (ASF) model. A sustained CO2RR activity – at least 12 h of continuous operation – suggested that, whichever the active sites were, they were continuously available at the catalyst surface over time. With the feeling of exploring uncharted territory, we devised strategies to understand how these unconventional nickel sites look and how they work.
Unveiling features of these active nickel sites drove our efforts for over one year in which we focused mainly on nickel phosphate, the most selective INO toward C2+ products. Our first idea was to identify the special nickel sites with undercoordinated ones. Computational simulations and the poor performance of defective Ni (average Ni coordination number of 7.5 vs 12.0 for the metallic phase) readily falsified it. After that, the unconclusive information we gathered from ex-situ characterization made us realize it was necessary to observe INOs in action. Operando X-ray absorption near-edge spectroscopy (XANES) was instrumental. Nickel phosphate-derived catalyst surprisingly retained stable positively charged Ni (Niδ+) sites (12%) at –1.2 V vs RHE. Operando extended X-ray absorption fine structure (EXAFS) linked them afterwards to Ni-O bonds. We obtained the same behavior from the borate-derived catalyst, suggesting a general feature of INOs (Fig. 1, right). We realized that the resistance to reduction of INOs was surprisingly high compared to the widely studied oxidic copper materials. After confirming that INOs catalysts bind CO more weakly than metallic Ni, a result deduced from CO stripping voltammetry experiments, we could finally claim that polarized nickel sites are the active ones.
At this stage, we decided to explore the first steps of the mechanism leading to such a variety of compounds. We first wondered which species could be the key initial intermediates. Co-feeding of different reactants, i.e., CO2, CO, formaldehyde, and formic acid, led to different performances. More precisely, CO2/CO and CO2/CH2O mixtures displayed ca. 3x and 2x formation rates compared to pure CO2. Another key difference with copper became thus evident: nickel allows C-C coupling pathways beyond CO dimerization, which is the accepted pathway on copper (Fig. 1, left).7 We identified intermediates directly formed from CO2, CO, and CH2O as key early pieces in the C-C coupling steps on INOs electrocatalysts. It was time for density functional theory to sketch a plausible mechanism able to incorporate all experimental observations: long hydrocarbons, polarized Ni sites, weak CO binding, and intermediates from CO2, CO, and CH2O for C-C coupling. Simulations on Ni(100) p(3×3) supercells with tunable degrees of polarization immediately rationalized how surface polarization prevents CO poisoning and enables the formation of C3+ hydrocarbons.8 The mechanistic uniqueness of polarized nickel sites is that adsorbates’ binding energy inversely correlates with the polarization degree and adsorbate dangling bonds, phenomenon coined as polarization-driven thermodynamic Linear Scaling Relationships9 (qLSR). This translates into a large multiplicity of precursors for C-C coupling beyond CO at different polarization degrees. We also predicted that reduced species, such as COOH and CH/CH2 can favorably couple on INOs catalyst surfaces to make the first C-C bond. What happens next? Further insertions of CH/CH2 sustain a mechanism to form C3+ hydrocarbons with a flavour reminiscent of the Fischer-Tropsch synthesis.
We have just started exploring the potential of INOs. We also trust that many of the concepts that we learned from this work could be translated to other materials. Production of synthetic fuels from CO2 at ambient conditions in one step is no longer utopic. It has just become a scientific challenge.
For more information, please read our article in Nature Catalysis at https://www.nature.com/articles/s41929-022-00803-5.
References
- Galán-Martín, Á. et al. Sustainability footprints of a renewable carbon transition for the petrochemical sector within planetary boundaries. One Earth 4, 565-583 (2021).
- Garcia de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm–2. Science 367, 661-666 (2020).
- Hori, Y., Murata, A., Takahashi, R. & Suzuki, S. Electroreduction of CO to CH4 and C2H4 at a copper electrode in aqueous solutions at ambient temperature and pressure. J. Am. Chem. Soc. 109, 5022–5023 (1987).
- Torelli, D. A. et al. Nickel–gallium-catalyzed electrochemical reduction of CO2 to highly reduced products at low overpotentials. ACS Catal. 6, 2100-2104 (2016).
- Calvinho, K. U. D. et al. Selective CO2 reduction to C3 and C4 oxyhydrocarbons on nickel phosphides at overpotentials as low as 10 mV. Energy Environ. Sci. 11, 2550-2559 (2018).
- Kudo, A., Nakagawa, S., Tsuneto, A. & Sakata, T. Electrochemical reduction of high pressure CO2 on Ni electrodes. J. Electrochem. Soc. 140, 1541-1545 (1993).
- Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732-745 (2019).
- Dattila, F. eFTS nickel. ioChem-BD https://doi.org/10.19061/iochem-bd-1-200 (2021).
- Pérez-Ramírez, J. & López, N. Strategies to break linear scaling relationships. Nat. Catal. 2, 971-976 (2019).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in