Loss of DOCK2 potentiates Inflammatory Bowel Disease–associated colorectal cancer via immune dysfunction and IFNγ induction of IDO1 expression
Published in Cancer and General & Internal Medicine
Why look at Inflammatory Bowel Disease-associated colorectal cancer (IBD-CRC)?
Inflammatory Bowel Disease-associated colorectal cancer (IBD-CRC) is a known and serious complication of Inflammatory Bowel Disease (IBD) affecting the colon. However, in comparison with its sporadic cancer counterpart, relatively little is known about its pathogenesis, and it has been the subject of much interest amongst gastroenterologists and cancer researchers alike.
IBD-CRC occurs on a background of colonic inflammation, occurs in younger patients, and is associated with increased mortality compared to sporadic colorectal cancer [1]. In addition, the mutational spectrum of IBD-CRC is very different from sporadic colorectal cancer, with alterations in TP53 being the most common driver event [2]. Most countries survey IBD patients for dysplasia or cancer at regular intervals after a diagnosis of Inflammatory Bowel Disease (IBD), a programme which has significant cost and service implications, particularly as the prevalence of IBD is soon estimated to approach 1 in 100 [3]. There is therefore an urgent need to understand IBD-CRC in greater detail, in the hope that greater understanding will lead to earlier diagnosis and improved treatment outcomes.
What led you to look at immune dysfunction in IBD-CRC?
Immune dysfunction has a significant part to play in the pathogenesis of IBD, as evidenced by the large number of immune-altering advanced therapies in the gastroenterologist’s armamentarium. Dedicator of cytokinesis 2 (DOCK2), an activator of the RAC pathway, is involved in both the innate and adaptive immune responses and was recently identified as a key driver gene in human IBD using network predictive modelling [4].
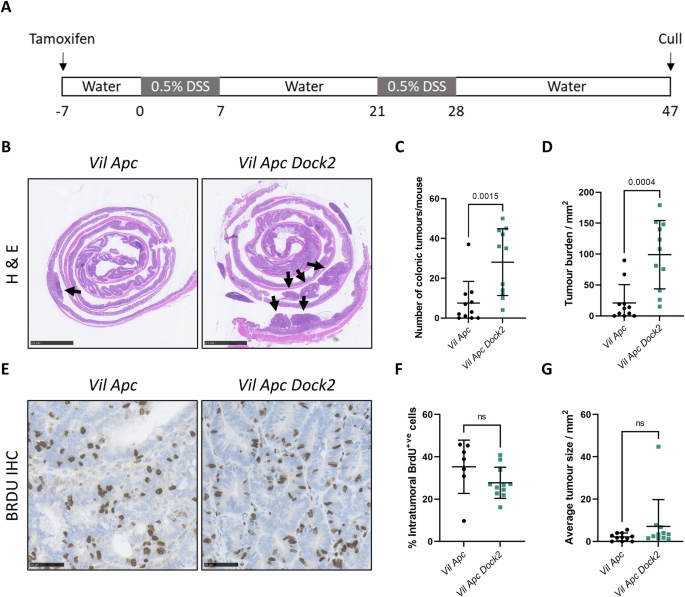
Here, we showed that loss of Dock2 in a Dextran Sodium Sulphate (DSS) colitis-induced mouse model of IBD-CRC increases tumour formation. Interestingly, when we examined the tumours from Dock2-deficient mice, they were infiltrated with higher numbers of CD3+ T cells. It was this initial finding that led us to look at immune pathways in more detail.
Subsequently, we undertook gene set enrichment analysis following RNA sequencing of these tumours. This showed increased interferon gamma signalling. We examined IFNγ target genes, and one of the most significantly upregulated was indoleamine 2, 3-dioxygenase 1 (Ido1). Ido1 is a previously described target of IFNγ signalling with known tumour promoting, immunomodulatory functions [5].
What did you find out about the mechanism underlying this immune dysfunction?
Having already demonstrated an increased CD3+ T cell population in tumours from mice lacking Dock2, further investigations of different T cell subtypes demonstrated that whilst CD4+ and CD8+ T cell infiltration was the same between groups, there were more γδ T cells than in control tumours. Flow cytometry analysis showed that a higher proportion of both γδ T cells and CD8+ T cells expressed IFNγ in Dock2 deficient colons compared to controls. We found that the majority of IFNγ producing cells were CD3 positive but IFNγ was also produced by CD3 negative cells, and the proportion of these cell was also significantly higher in Dock2 deficient mice. Together, this suggests Dock loss leads to immune dysregulation, characterized by increased IFNγ production in multiple immune subtypes including, but not restricted to, γδ T cells and CD8+ T cells.
We then co-stained tumour tissue from mice lacking Dock2 for epithelial and immune cell markers alongside IDO1. IDO1 expression was exclusively co-localised with CDH1 expression, and we did not observe co-expression with CD3 or CD8 markers. This suggested an induction of IFNγ signalling in the tumour epithelium of Dock2 deficient mice. We also found that IDO1 protein and mRNA expression was induced in a dose dependent manner by increasing concentrations of IFNγ in tumour colonic epithelial organoids.
IDO1 is a tryptophan catabolic enzyme that catalyses the conversion of tryptophan to kynurenine and other downstream metabolites. Consistent with the elevated expression of IDO1, we observed a depletion of tryptophan following Dock2 deletion, when analysed using mass spectrometry.
Finally, to test the functional significance of elevated IDO1 expression and tryptophan metabolism in Dock2 deficient tumours, we examined the effect of IDO1 inhibition on tumourigenesis. We treated Dock2 deficient mice with the IDO1 inhibitor 1-L-MT whilst undergoing repeated rounds of DSS induced colitis. Mice treated with 1-L-MT had a significantly lower tumour number and tumour burden compared to mice treated with vehicle. Importantly, this was not due to IDO1 inhibition impacting on severity of colitis, confirming the tumour promoting effects of increased epithelial IDO1 activity following Dock2 deletion.
What’s the clinical relevance of these findings?
We investigated the expression of the IFNγ target IDO1 in a set of human samples encompassing different stages of IBD-CRC disease progression. IHC analysis showed that IDO1 expression is significantly increased in inflamed tissue compared to normal colon and this increased expression persists through to the development of cancer. We also found that DOCK2 mutation and/or low DOCK2 expression correlates with poor prognosis in microsatellite stable (MSS) CRC patients, a group that is generally immune supressed, further supporting a tumour suppressor role for this protein.
What do we still not know about the proposed mechanism?
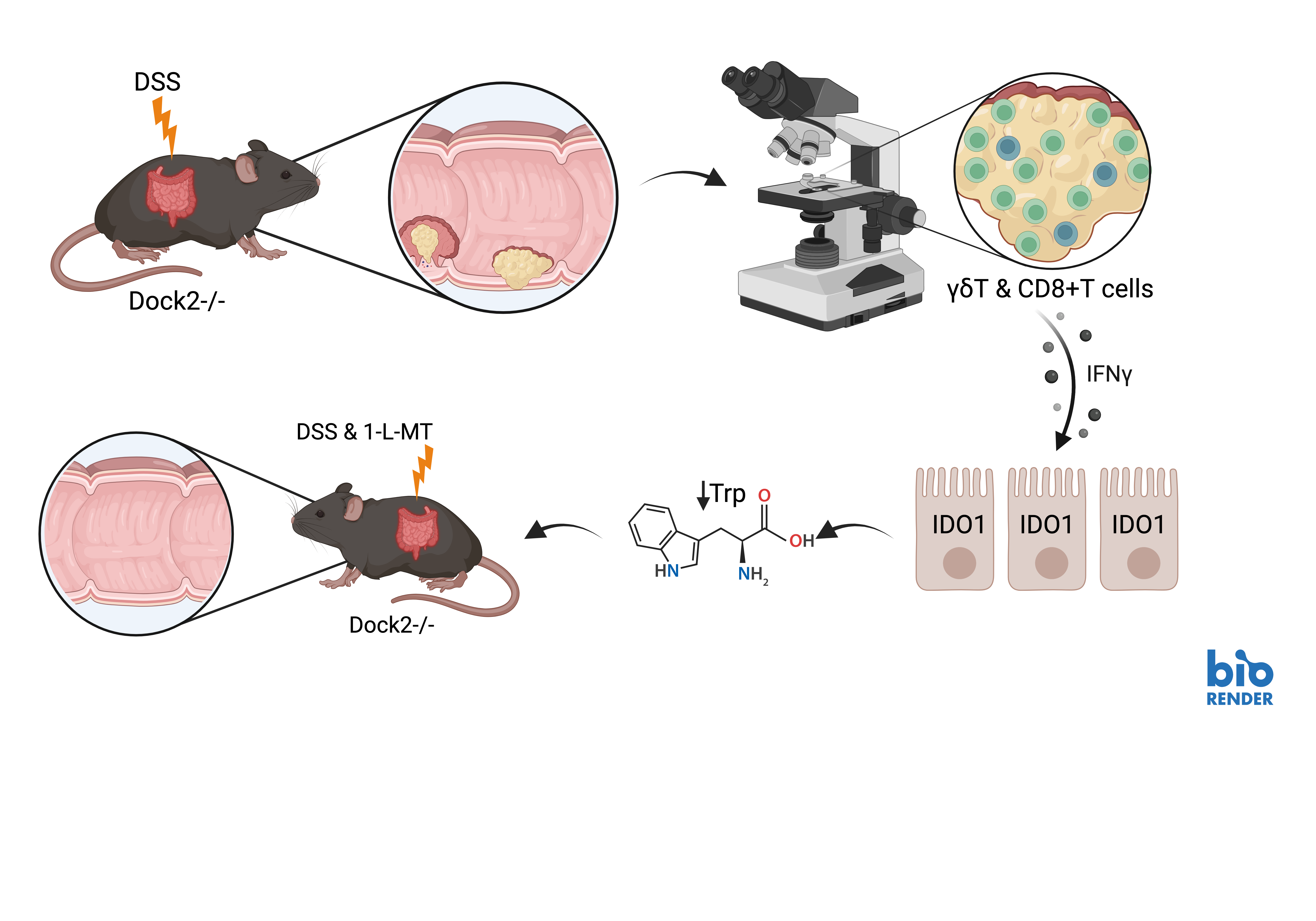
Taken together, these results suggest that loss of Dock2 leads to immune dysregulation, enhancing tumourigenesis via interferon gamma induced expression of IDO1. In mice lacking Dock2, IFNγ production is increased in multiple T cell populations, including CD8+ and γδ T cells, and administrating IFNγ to tumour organoids induces robust IDO1 expression suggesting this response is driven by immune cell IFNγ production. Finally, treatment with 1-L-MT, an IDO1 inhibitor, abrogates the increased tumourigenesis observed in mice without Dock2.
It it is not clear, however, why more IFNγ producing T cells accumulate in tumours formed in Dock2 mutant mice, a gene that is expressed primarily in hematopoietic cells. These cells may be recruited to tumours by increased expression of chemokines, or they may proliferate in situ. Neither is it clear why a predominance of IFNγ-producing T cells, normally associated with anti-tumour activity [6], correlate with increased tumourigenesis.
Despite these questions, we have highlighted the intricate role of the immune and interferon gamma response in IBD-CRC. This enhances our understanding of the aetiology of IBD-CRC and identifies these processes as potential therapeutic targets for this complex and chronic disease.
Figure 1: Schematic of Dock2 loss, leading to increased tumourigenesis, induced immune dysregulation, interferon γ upregulation and IDO1 induction, with phenotypic rescue following IDO1 inhibition. Created with BioRender.com.
References:
- Jewel Samadder N, Valentine JF, Guthery S, Singh H, Bernstein CN, Wan Y et al. Colorectal Cancer in Inflammatory Bowel Diseases: A Population-Based Study in Utah. Dig Dis Sci 2017; 62: 2126-2132.
- Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016; 150: 931-943.
- Jones GR, Lyons M, Plevris N, Jenkinson PW, Bisset C, Burgess C et al. IBD prevalence in Lothian, Scotland, derived by capture-recapture methodology. Gut 2019; 68(11):1953-1960.
- Peters LA, Perrigoue J, Mortha A, Iuga A, Song WM, Neiman EM et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet 2017; 49: 1437-1449.
- Bishnupuri KS, Alvarado DM, Khouri AN, Shabsovich M, Chen B, Dieckgraefe BK et al. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res 2019; 79: 1138-1150.
- Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med 2003; 198: 433-442.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in