m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome.
Published in Bioengineering & Biotechnology
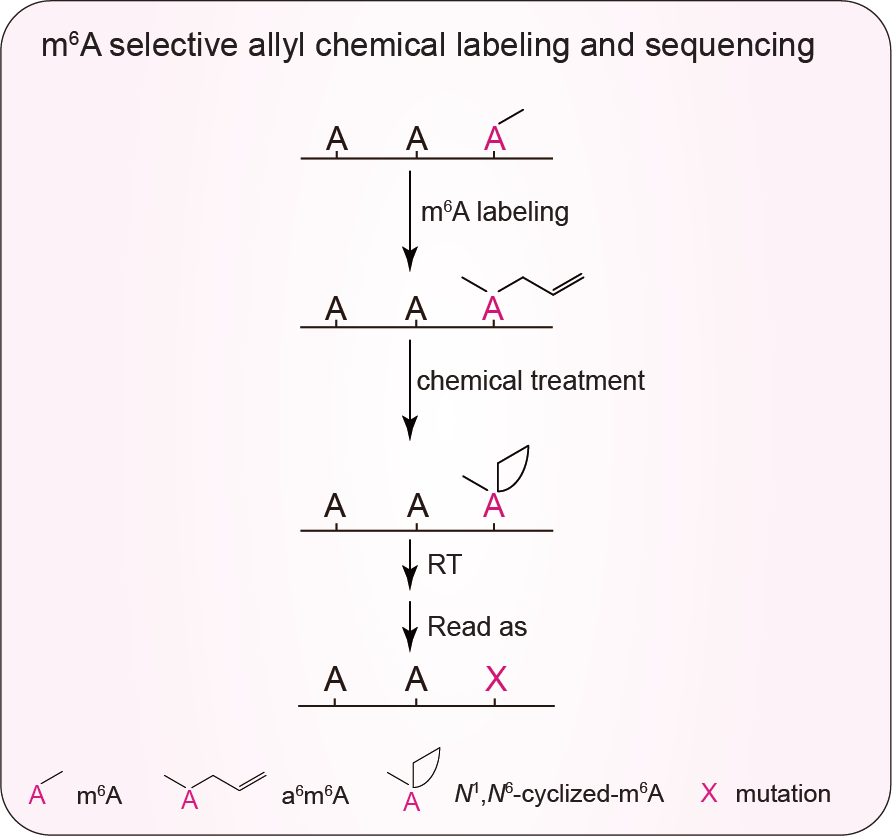
We report a quantitative m6A-SAC-seq method that maps m6A sites in the entire transcriptome at single-base resolution with the modification stoichiometry information. m6A-SAC-seq enables dissecting dynamics and functional roles of m6A sites in diverse biological processes using limited input materials.
The antibody-based m6A MeRIP-seq profiling approach has been widely adopted and has enabled numerous discoveries of m6A biology; however, the antibody-based approach has several limitations, including low resolution, a lack of stoichiometric information, a requirement for a large amount of input materials, and a limited ability to compare m6A methylation level across conditions. Other methods such as DART-seq 1, MAZTER-seq 2and m6A-REF-seq 3 have also been developed; however, none of these current methods provides whole transcriptome detection of m6A at base-resolution with modification stoichiometry information. Unlike in studies of DNA 5mC methylation in which bisulfite sequencing serves as a gold standard, we currently do not have a quantitative method to map m6A in the entire transcriptome. This technological bottleneck, coupled with the requirement of a large amount of input materials in most available methods, significantly hamper our ability to study m6A biology, especially during cell differentiation and tissue development as well as with clinical samples.
I joined Professor Chuan He’s laboratory at University of Chicago in February, 2015 and initiated my first project on new method development of DNA 5hmC sequencing at near-base resolution4. In 2018, Professor He introduced this more challenging direction on developing single-base resolution sequencing technology for mapping RNA m6A. The original idea was to employ the ADAR deaminase enzyme to deaminate all unmethylated adenosines in the transcriptome. However, we encountered numerous challenges as the RNA secondary structure preference and motif selection of ADAR make the identification of m6A sites a daunting task. Therefore, we turned our attention to positively detecting m6A.
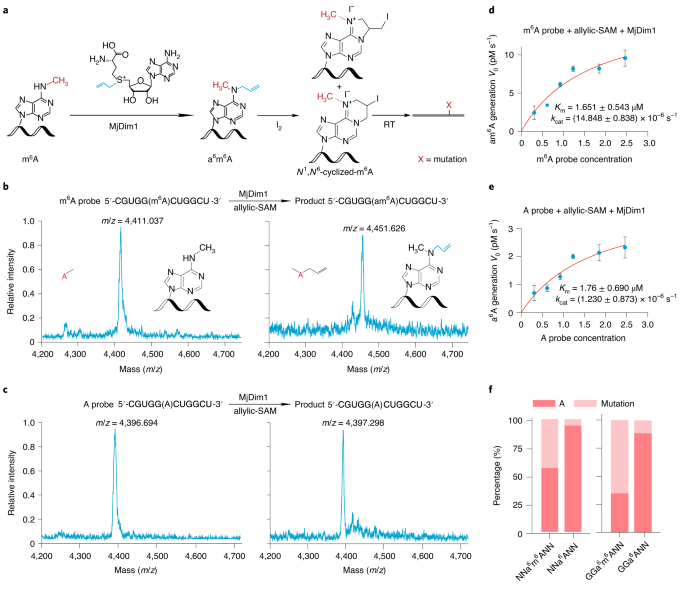
After extensive literature reading and experimental trials, we selected the Methanocaldococcus jannaschii homolog MjDim1, a member of the Dim1/KsgA family of dimethyl transferases, which transfer the methyl group from S-adenosyl-L-methionine (SAM) to adenosines, forming m6A and then N6, N6-dimethyl-adenosine (m62A) in consecutive methylation reactions5. MjDim1 showed highly processive kinetics of converting m6A into m62A6, and employed a chemically modified allylic-SAM as the co-factor7 (Fig. 1). In the presence of allylic-SAM, MjDim1 exhibited a notable, approximately 10-fold preference for m6A over A in a model allyl group transfer reaction, converting m6A into allyl-modified m6A (N6-allyl, N6-methyl adenosine or a6m6A), and “A” into allyl-modified “A” (N6-allyl adenosine or a6A).
We further optimized enzyme labeling protocol to achieve the optimum efficiency. Subsequent I2 treatment converts a6m6A and a6A into homologs of N1, N6-ethanoadenine and N1, N6-propanoadenine, respectively, as we have previously shown7 (Fig. 1). The HIV-1 RT reads through the allyl-labeled and cyclized adducts with negligible RT stops. The HIV-1 RT generated ~10-fold higher mutation rates at the cyclized a6m6A sites (true positive m6A sites) than cyclized a6A sites (unmodified A sites). Therefore, m6A-SAC-seq directly detects m6A and exhibits high selectivity towards m6A over A at two steps of the procedure: i) the allyl transfer from allylic-SAM catalyzed by MjDim1 is ~10-fold more selective for m6A than A; and ii) the labeled and cyclized m6A adducts generate higher mutation rates than the corresponding adducts formed from unmodified A. Therefore, we can achieve ~100-fold selectivity towards m6A over A in m6A-SAC-seq. To confirm the specificity of m6A-SAC-seq, we could include a control sample in which RNA is treated with the m6A demethylase FTO before MjDim1 labeling to ensure detection of m6A sites. This step is not necessary using our latest computational pipeline.
After optimization, we can apply m6A-SAC-seq using ~30 ng of input RNA, making it suitable for investigating a variety of biological processes. We obtained the first comprehensive maps of m6A at single-base precision and with stoichiometry information. Numerous cell-type-specific m6A sites were observed in RNAs isolated from HeLa, HEK293 and HepG2 cells by clustering stoichiometry information, with quantitative m6A fractions of a large portion of m6A sites differing among these different cell types. This observation is consistent with the notion that certain cell-type specific transcriptional factors could recruit the methyltransferase complex to deposit m6A for the regulation of cell-type specific functions 8-10.
While the RNA m6A modification plays critical roles during cell differentiation, tissue development and transcriptional regulation, studies of these biological processes have been hampered by the limited materials for sequencing and lack of robust methods for comparison of m6A levels across conditions. We further employed m6A-SAC-seq to investigate human umbilical cord blood derived CD34+ HSPC differentiation into monocytes as an example (Fig 2). We mapped m6A at base-resolution with stoichiometric information at four different time points along HSPC differentiation. We observed a highly dynamic picture of m6A fraction changes at all regions of mRNA during differentiation; hundreds of gained or lost m6A sites were observed in the 5’ UTR, CDS, 3’ UTR and intronic regions, respectively, between any of the two time points. Notably, transcripts of key transcriptional factors as well as mRNAs regulated by these transcriptional factors tend to be dynamically m6A methylated during HSPC differentiation, and their transcript level changes tend to correlate with differential m6A methylation, suggesting that the m6A methylation is used by cells to prime differentiation. These quantitative m6A maps thus serve as important resources and starting points for future mechanistic investigations and potential manipulation of HSPC differentiation through modulating m6A stoichiometries on selected transcripts.
Our study showcases the potential for broadly applying m6A-SAC-seq to obtain whole-transcriptome m6A stoichiometry changes in various cell differentiation, early development, neuronal signaling and clinical samples. We think m6A-SAC-seq will serve as a gold standard that overcomes the current technological bottleneck for quantitative m6A sequencing and enables new biological discoveries.
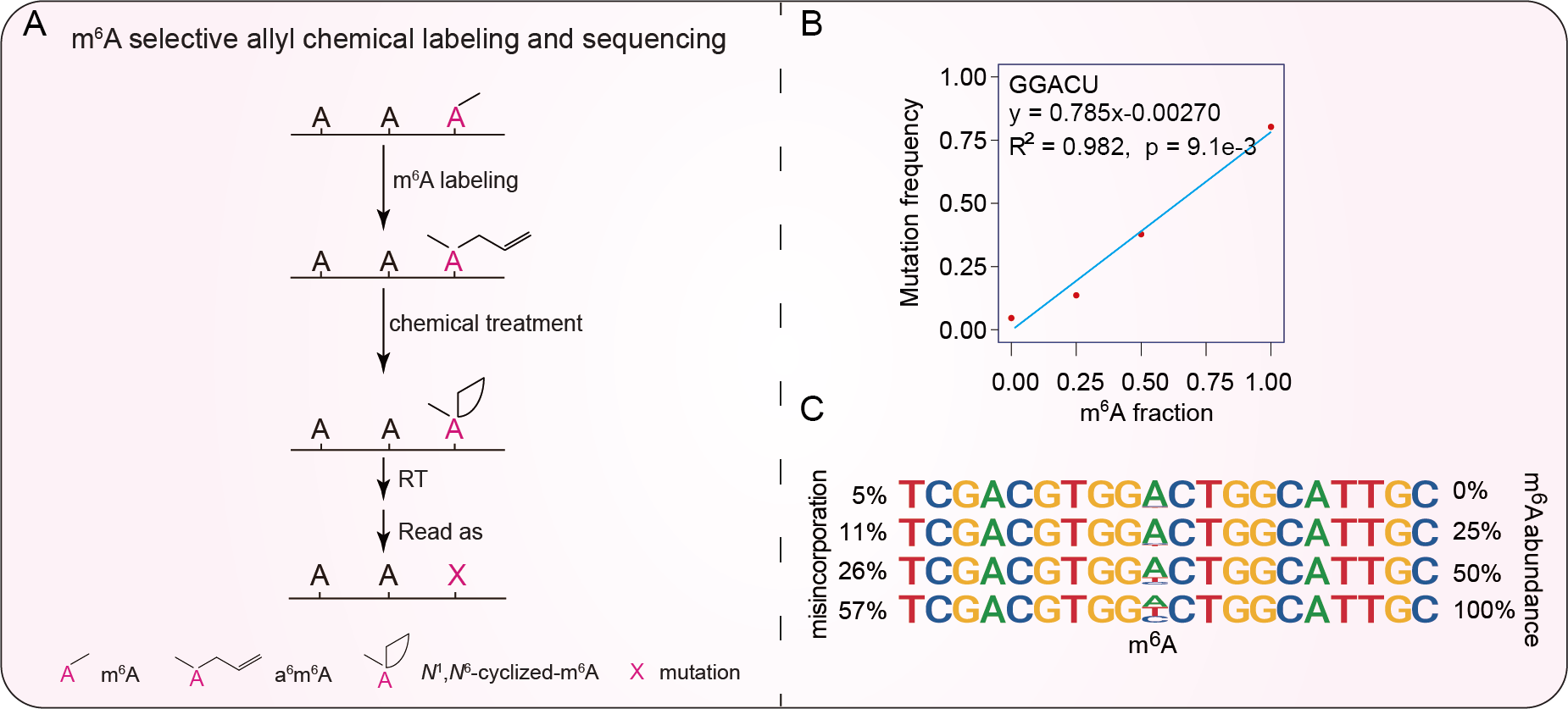
Fig. 1 m6A-SAC-Seq strategy and development.
Schematic diagram of m6A-SAC-Seq. A: MjDim1 uses allylic-SAM as a co-factor to label m6A to allyl6m6A, which undergoes cyclization upon I2 treatment. Cyclized a6m6A induces mutation using HIV reverse transcriptase during reverse transcription. B: Calibration curve for each GG(m6A)CU motif is generated by linear regression. P-values and goodness of fit (R2) of linear regression were also shown. C: Examples of mutation pattern of an m6A-modified 41-bp spike-in probe with 0%, 25%, 50%, 100% of m6A, respectively.

Fig. 2 m6A Dynamics across hematopoietic stem cell differentiation into monocytes.
Base-resolution m6A dynamics with stoichiometric information at four different time points along HSPC differentiation.
- Meyer, K.D. DART-seq: an antibody-free method for global m6A detection. Nat. Methods. 16, 1275-1280 (2019).
- Garcia-Campos, M.A. et al. Deciphering the "m6A Code" via antibody-independent quantitative profiling. Cell 178, 731-747 e716 (2019).
- Zhang, Z. et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv 5, eaax0250 (2019).
- Hu, L. et al. Jump-seq: Genome-Wide Capture and Amplification of 5-Hydroxymethylcytosine Sites. J Am Chem Soc 141, 8694-8697 (2019).
- O'Farrell, H.C., Musayev, F.N., Scarsdale, J.N. & Rife, J.P. Binding of adenosine-based ligands to the MjDim1 rRNA methyltransferase: implications for reaction mechanism and drug design. Biochemistry 49, 2697-2704 (2010).
- O'Farrell, H.C., Pulicherla, N., Desai, P.M. & Rife, J.P. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA 12, 725-733 (2006).
- Shu, X. et al. N6-allyladenosine: a new small molecule for RNA labeling identified by mutation assay. J. Am. Chem. Soc. 139, 17213-17216 (2017).
- Zhang, Z. et al. Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat. Genet. 52, 939-949 (2020).
- Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126-131 (2017).
- Bertero, A. et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 555, 256-259 (2018).
My background includes structural, biochemical, molecular and cellular biology. I have extensive experience in protein purification, biochemistry and molecular and cellular experiments. My skills include a comprehensive background in epigenetics, DNA modification, structural biology, gene regulation, Next generation sequencing and DNA/RNA epigenetic modification detection method development. I am an expert in determining enzymatic properties, protein purification, molecular and cellular operations including cell culture, immunoprecipitation, Chip-seq, DNA synthesis and purification etc.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in