Making nanopore sequencing and de novo assembly fast and efficient
Published in Bioengineering & Biotechnology


Article: Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes
When choosing a strategy for genome sequencing, scientists face two common choices: "Reference-based analysis or de novo assembly?" and “Long-reads or short-reads?”
Unsurprisingly, most choose reference-based methods using short-reads because they are fast, cheap, easy and proven.
However, the devil is definitely in the details. Short-reads alone fail to generate contiguous assemblies of large genomes because the reads are simply too short1. Using existing scaffolds of an existing reference, short-read can accurately find most small variations2. Although finding small variations made short-reads the genomic workhorse, variant-identification only works in the unique portions of the reference genome, leaving out duplicated and repetitive sequences2. Also, reference-based methods naturally detect alleles similar to the reference, introducing reference allele bias3. This bias is strong for structural variations, which are often missed or miscalled. Short reads often can’t span neighboring variations, so while genotypes are ascertained, the phasing relationships (allelic organization along the maternal and paternal chromosomes) are not4. Until recently long-read de novo assembly methods were expensive, time-consuming, and generally reserved for new species. We asked ourselves: can these challenges be overcome using long-reads and cheap, fast, and scalable methods?
In 2014, the phone-sized, low-cost, Oxford Nanopore MinION was released which democratized third-generation sequencing by making it accessible. In 2017, we participated in an international effort that produced the first reported de novo assembly of a human genome (HG001) using nanopore sequencing. This effort used 53 MinION flowcells, 150,000 CPU hours, and weeks of wall-clock time. While promising, this long-read assembly approach had extensive computational and sequencing requirements5.
In 2018, the high-throughput Oxford Nanopore PromethION device was released. To assess its performance, we sequenced eleven human genomes in nine days and achieved 60x coverage (with ~7x coverage in 100kb+ reads) per sample. This unprecedented sequencing speed required improvements in genome assembly methods. Contemporary approaches for nanopore data-based assemblies required one week of wall-clock time and cost ~$1000.
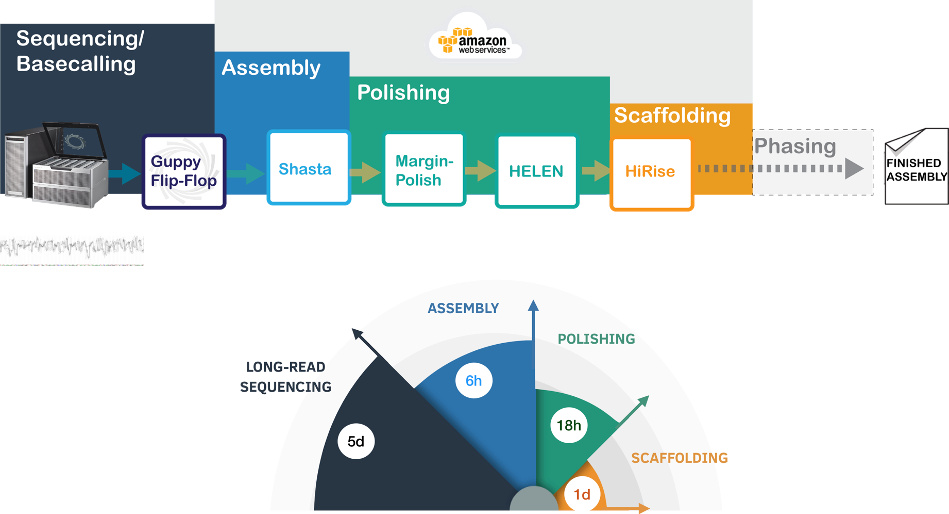
Figure 1: Overview of the pipeline for rapid de novo assembly. The sequencing is performed on the PromethION sequencing device that can run 48 flow-cells in parallel. Shasta can generate and assemble a human scale genome under 6 hours and MarginPolish-HELEN can polish within 18 hours. Finally, scaffolding using proximity ligation (Hi-C) data takes 24 hours. Overall, we can generate de novo assemblies of a large cohort of human genomes efficiently which will allow us to study variations in the human genome in an unbiased manner.
In collaboration, with CZI’s Paolo Carnevali, we developed Shasta, a fast and accurate genome assembly tool. We show Shasta can routinely yield a high-contiguity human genome assembly within 6 hours, costing ~$70 on an AWS instance! Additionally, Shasta not only has comparable (or better) base-level assembly accuracy as its contemporaries but also has the lowest number of misassemblies. To further improve base-level quality, we developed a deep neural network-based consensus sequence polisher, MarginPolish-HELEN. The total cost of Shasta-MarginPolish-HELEN is under $200 and 37 hours in wall-clock time, reducing the overhead of generating long-read assemblies by a factor of five. Finally, we used proximity ligation information from Hi-C sequencing to achieve near
chromosome-level scaffolds. We successfully applied this strategy to all eleven genomes.
To orthogonally validate assembly quality, we used CHM13 (haploid sample) nanopore data from the telomere-to-telomere (T2T) consortium6. MarginPolish-HELEN produced Q30 or 99.9% accurate assembly using only nanopore data, a first for the human genome.
Our effort in making nanopore long-read de novo assembly easy, cheap, and fast will, we hope, permit a more comprehensive and unbiased assessment of human, plant and animal variation. We are now integrating phasing into the assembly and polishing pipeline. We anticipate achieving de novo assemblies that will be contiguous, complete, and phased over structural variants. Applied to large cohorts, this will reveal new insights into the harder to measure regions of the human genome.
References
1. Wong, K. H. Y., Levy-Sakin, M. & Kwok, P. Y. De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations. Nat. Commun. 9, 1–9 (2018).
2. Poplin, R. et al. A universal snp and small-indel variant caller using deep neural networks. Nature Biotechnology vol. 36 983 (2018).
3. Brandt, D. Y. C. et al. Mapping bias overestimates reference allele frequencies at the HLA genes in the 1000 genomes project phase I data. G3 Genes, Genomes, Genet. 5, 931–941 (2015).
4. Ebler, J., Haukness, M., Pesout, T., Marschall, T. & Paten, B. Haplotype-aware diplotyping from noisy long reads. Genome Biol. 20, (2019).
5. Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
6. Miga, K. H. et al. Telomere-to-telomere assembly of a complete human X chromosome. bioRxiv 735928 (2019) doi:10.1101/735928.
Follow the Topic
-
Nature Biotechnology

A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in