Making Sense of the Chaos of Collagen Self-Assembly
Published in Chemistry

When I try to explain this project in one sentence, it’s very difficult. My best attempts contain some version of the following phrases, “collagen is a protein that forms the foundations of the structures in your body,” “there are still a lot of things we don’t understand about collagen,” “I’m trying to use computers to better understand why collagen is so stable,” “if we understand collagen stability better, we can determine important sections of collagen, design materials that can mimic collagen, or design molecules that can interact with collagens.” However, when I joined the lab, I had the opportunity to present my research to a class of 7th graders, and while all of those phrases were important for effectively communicating the science, I had a captive audience I could teach some of the molecular details of my work as well.
I decided that the best, clearest, way to go about doing this was with a few presentation slides, a handful of pipe cleaners, and some beads. Admittedly, some of the scientific sophistication was lost with such a simplistic model, but it served the purpose of simplifying the discussion for my audience. Collagen proteins consist of three strands that twist around one another to form a triple helix. This was illustrated by handing three pipe cleaners to each 7th grader to twist into triple helices. Helices consisting of pipe cleaners of all the same color illustrated homotrimers whereas helices with mixed colors represented binary and ternary heterotrimers. Blue and red beads were used to represent amino acids and charge pair interactions. Red beads represented positively charged amino acids and blue beads represented negatively charged. With these, the 7th graders could design their own complimentary alignments of multiple “amino acids” for prompting their triple helices to “form” in a specific arrangement.
 Figure 1. Pipe cleaner triple helices. Top to bottom: white homotrimer, green and purple binary heterotrimer, white yellow and blue ternary heterotrimer, and yellow blue and red ternary heterotrimer with included charge pair interactions for controlling registration.
Figure 1. Pipe cleaner triple helices. Top to bottom: white homotrimer, green and purple binary heterotrimer, white yellow and blue ternary heterotrimer, and yellow blue and red ternary heterotrimer with included charge pair interactions for controlling registration.
While simplistic, this approximately represents the state of this project when I joined the lab: We understood the structure of collagens and how to use positively charged lysines paired with negatively charged aspartates to promote the formation of designed triple helices. We also had a computer algorithm that could assess the likelihood that the designed triple helix would fold as intended rather than competing assemblies, which is called specificity. As I started my PhD research, we began to set goals for improvements to the existing algorithm. Our first was to improve the algorithm’s ability to predict triple helices that will fold exclusively into one structure, because while it was accurate in its predictions we wanted to design triple helices with better specificity. Minor progress was possible with the existing set of amino acids, but it quickly became evident that a larger set would be necessary. So, our next goal was to include other amino acid interactions already used in the literature. In this process, due to the different effects of different amino acids on the stability of triple helices, it became apparent that the easiest and most precise way to predict helix specificity would be through predicting helix melting temperatures rather than predicting arbitrary stability values. We added this to our list of goals.
Around this time, because the coding and predicting aspects were taking so long, we began working on what we thought was a tangentially related project in which we mathematically calculated the stabilizing effect of both new and established pairwise interactions through synthesis and stability measurements of single and double substituted helices. We were excited to better understand and expand the selection of amino acid interactions for designing new triple helices known to the field.
Eventually, we realized that the two projects were more closely related than we had originally considered. The mathematical deconvolution supplied thermal stability values and interactions to the algorithm that were otherwise difficult to calculate or ascertain. The two were merged and with that as inspiration, we added a goal to assess all natural amino acids with our algorithm.
The remaining data we needed for the effects of single amino acid substitutions exists courtesy of the lab of Anton Persikov and the lab of Barbara Brodsky. Barbara Brodsky has been pivotal for expanding the field’s understanding of collagen folding and stability. So, it is no surprise that, in addition to many other peptides, all 41 iterations of homotrimers necessary to understand the effects of the natural amino acids on collagen structures had already been synthesized and analyzed. This provided a quick source for expanding the selection of amino acids in our algorithm.
However, the code required an overhaul. With the existing format, each new amino acid assessed increased the number of lines of code as a factorial of the total number of amino acids assessed. This rate of growth was an unsustainable pattern of increase. However, a simple loop iterating through all amino acids to correlate amino acid stabilities and two types of interactions was just not feasible with the existing set-up. So, as a mostly self-taught programmer with only four months of official Java training, I figured out using tools that computer programmers call HashMaps and String concatenation to work for what I needed. For the new format, every new amino acid added to the code only requires new lines to define the new coefficients used by the algorithm, the one new amino acid stability value and the new pairwise interaction values.
With the algorithm now assessing all natural amino acids, the problem arose in which the algorithm had to choose which available interaction to use for each amino acid in a peptide sequence. In a triple helix, each amino acid has two potential geometries of interaction, so it also has the choice between interacting with two different amino acids. The algorithm should choose the most stable combination of competing interactions, but two choices at each position creates an extending network of choices, each of which depends on the state of others in the network. The only way to address this problem was with a recursive algorithm: each choice is checked and compared to the others in the network until the most stable combination is determined. Recursive algorithms are classically tricky to write and more elegant than iterative algorithms. The day I got that recursive algorithm figured out, written, and working is one of my proudest moments from graduate school.
After that, it was time to name the algorithm. “Collagen-like peptides” and “collagen-mimetic peptides” have been used in the past to describe these systems, but neither CLP nor CMP lend themselves to easy acronym design, so I began to think of other possibilities and realized that “collagen-emulating peptides” (CEP) would work well in an acronym. The first of these attempts was ENCEPTSHUN3 (Estimator of N Collagen-Emulating Peptides’ Temperature of Significant Helical Unfolding for N<=3). While this was fun and sub-sub-subconsciously suggests a connection to a certain Christopher Nolan and Leonardo DiCaprio film, it also is a mouthful and a bit reaching. SCEPTTr (Scoring function for Collagen Emulating Peptides Temperature of Transition) flows more fluidly off the tongue, makes for a simple precise name, and just felt right.
SCEPTTr next needed to be trained to be more accurate and precise. The aforementioned method for determining amino acid interactions is a helpful starting point, but has limitations. 1) Understanding many interactions requires many syntheses. 2) Each interaction determined this way has an associated degree of error and uncertainty. A promising way to overcome these limitations is by utilizing existing data. There exist over 400 synthetic collagen triple helices in the literature. These triple helices possess a large sampling of pairwise interactions that can be considered and incorporated into SCEPTTr and they possess multiple iterations of some interactions which can help hone the precision thereof and enable SCEPTTr to make more accurate predictions. This library of over 400 triple helices also supplies a suite of testable values with which to assess SCEPTTr’s predictions.
The utilization of the library for both these applications enables training the algorithm values with a machine learning algorithm. I wrote a genetic algorithm that adjusts the scoring values to optimize the performance of SCEPTTr’s predictions of the library triple helices. Applying this genetic algorithm increased the R2 of the prediction from ~0.65 to ~0.95 which was so satisfying.
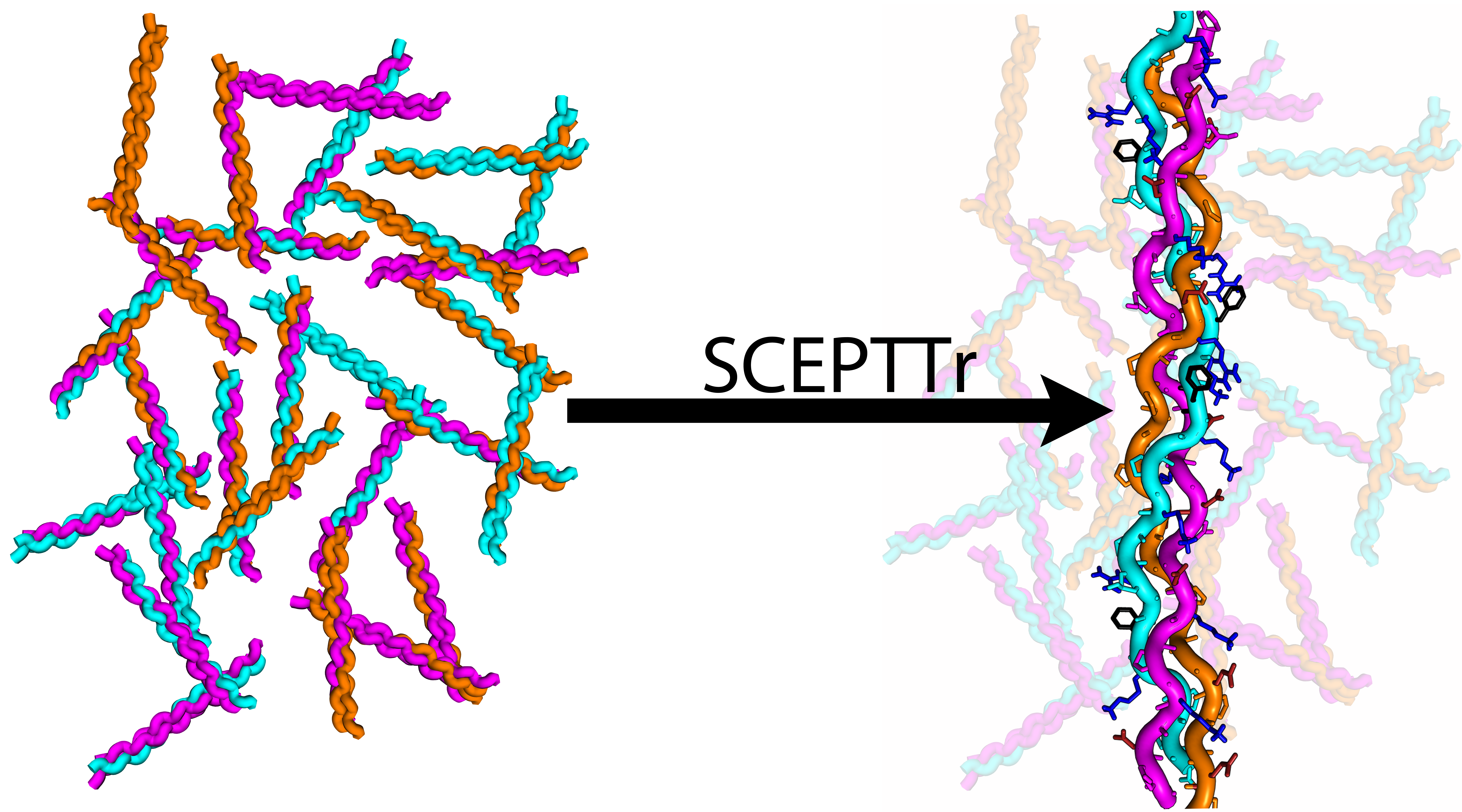 Figure 2. Visualization of the utility and benefit of SCEPTTr for predicting triple helical stability. SCEPTTr assesses the stability of all possible triple helices and predicts the identity of the species which folds in solution.
Figure 2. Visualization of the utility and benefit of SCEPTTr for predicting triple helical stability. SCEPTTr assesses the stability of all possible triple helices and predicts the identity of the species which folds in solution.
This level of improvement and the increased number of understood amino acid interactions finally allowed revisiting the initial goal. We designed a new triple helix, synthesized all of the peptides, and characterized the resulting triple helices. This combination of peptides resulted in a new stable triple helix that assembles as designed, which was very encouraging! While this triple helix is stable and the competing structures are unstable at room temperature, some of the competing species still fold and melt just below room temperature. This combination of features partially achieved our goal, but leaves room for improvement in the future, as always.
By the time of this publication, we have increased the sophistication, the scope, the accuracy, and the precision of our prediction algorithm in ways that will be highly beneficial for the field. Funny enough though, even with all of these improvements, for the sake of effective communication, perhaps the best way to explain the project to a class of 7th graders is still with a handful of pipe cleaners and some beads. And honestly, explaining scientific concepts to 7th graders—encouraging them to pursue science—is something I see as just as important as, if not more than, the rest of the work I have done and will do as a scientist.
Original paper: https://doi.org/10.1038/s41557-020-00626-6
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in