Measuring RNA modifications with evolved enzymes
Published in Protocols & Methods

When I started my lab at the University of Chicago five years ago, the very first project we initiated in my then new group was an effort to engineer RNA polymerases (RNAPs) as a new family of biosensors. The idea was the biosensors that encode molecular information in genetic outputs could be useful new tools for synthetic biology. After showing the concept was feasible (https://pubs.acs.org/doi/abs/10.1021/jacs.5b10290) we then developed an elaborate evolution system to develop RNAPs into a versatile biosensor platform (https://www.nature.com/articles/nchembio.2299). Ultimately, these new tools allowed us to move into a variety of new areas in synthetic biology and serve as one of the focal points of my group.
Our work with RNAP-based biosensors taught us one thing: polymerases are incredibly complex molecular machines! The way mutations correlated with functional changes in the protein were very hard to predict or even rationalize after the fact - which is why our unbiased evolutionary approaches were effective design principles to solve challenges with polymerase engineering.
Around three years ago my colleague, office-mate, lab neighbor, and friend Professor Chuan He came to me with a problem. He had been trying to study an exciting new mRNA chemical modification, m1A, but was having lots of problems with the one available reverse transcriptase (RT) enzyme capable of detecting it. Given my group's background in polymerase engineering in general, perhaps we could devise a better enzyme to tackle this challenge.
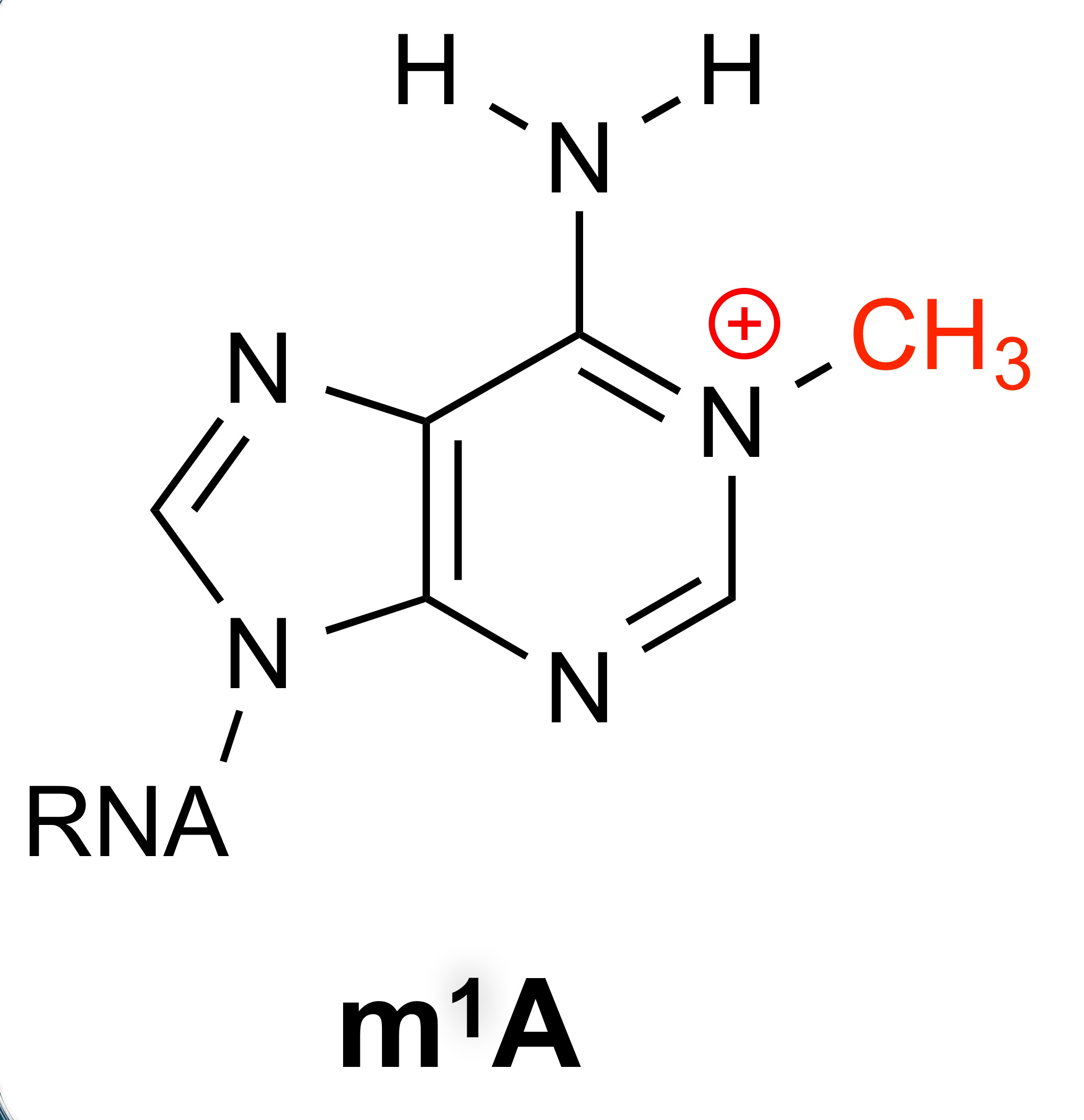
From a chemistry perspective, I was very excited about the idea of studying m1A. Unlike other RNA modifications, such as m6A, which are rather minimally perturbative, m1A is a very perturbative modification. Not only does the methylation make the base cationic (so quite perturbative from a physical organic perspective), but the modification is also right at the Watson-Crick-Franklin base pair interface, disrupting canonical base pair interactions. Therefore, I was sold that this was an exciting area of research. Chuan and I recruited a very talented new post doc, Dr. Huiqing (Jane) Zhou, to come work jointly in our groups to tackle this problem.
Given our experiences with RNAP engineering, we immediately recognized that the only way we were going to have success was to leverage a rapid screening/evolutionary approach to tackle this challenge. We decided to develop an in vitro fluorescent screen for RTs. The reason m1A is such a difficult modification to detect is that RTs in general cannot read through the modofication, resulting in the generation of truncated products. Therefore, we devised a screen, based on a fluorescent aptamer developed by Professor Samie Jaffrey -(https://pubs.acs.org/doi/abs/10.1021/ja508478x) that yields a fluorescent signal if and only if an RT can both read-through and selectively mutate at an m1A site.
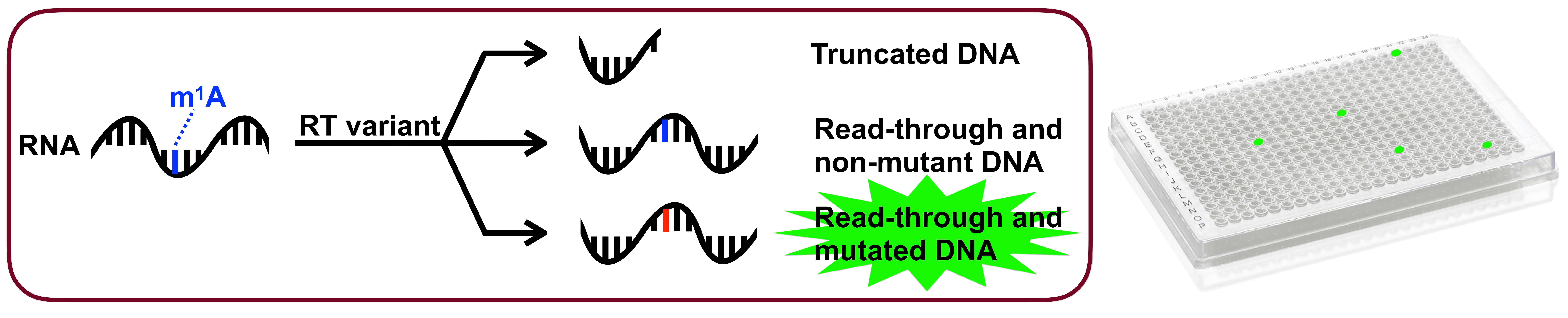
We decided to start our engineering efforts with HIV RT - a robust enzyme with lots of properties that make it well-suited to biotechnology applications and sequencing, but with almost no ability to read-through m1A. A given variant of HIV RT, therefore, would primarily generate truncated DNA product. Our screen would provide fluorescent signal if mutations we screened through in the enzyme would allow it to read-through and selectively mutate.
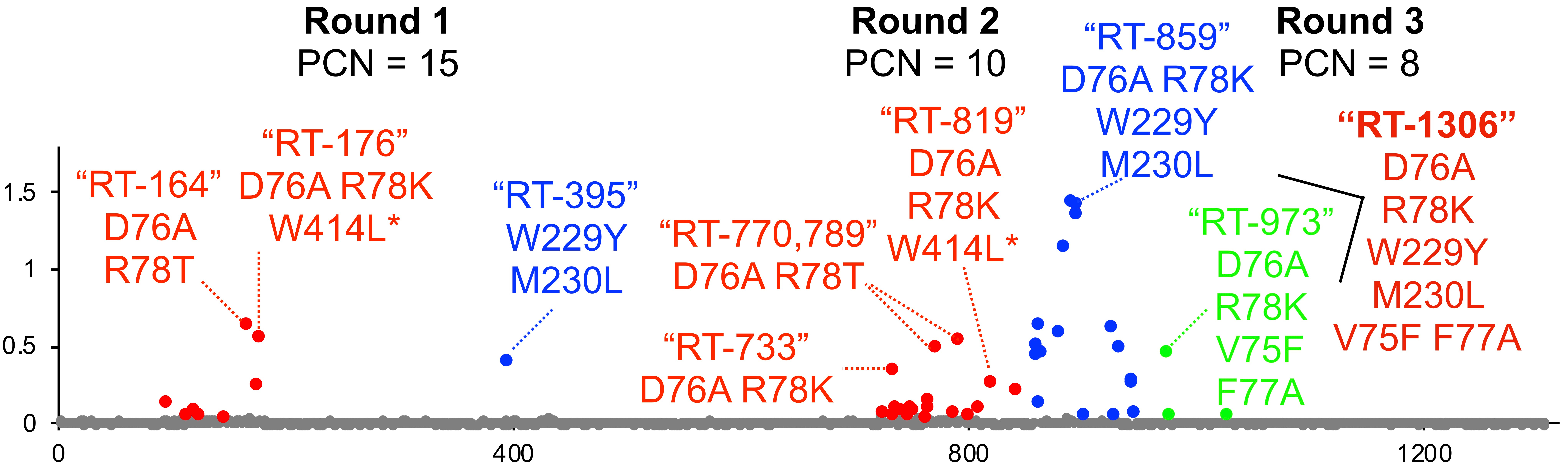
Once we optimized the screen, we generated libraries of HIV RTs to test. We used structural information to target mutations around the active site, assuming that is where we would find success. Out of the first ~700 variants we assayed, almost all (>95%) were inactive. Fortunately, we found a few variants with a sliver of activity. In directed evolution, often times once you find your first small success, you can use that activity to pry open broader activity. Indeed, it only took a few additional rounds of screening to ultimately discover "RT-1306" - named such because it was the 1,306 enzyme we screened through! This really highlights the throughput of our new screening method.
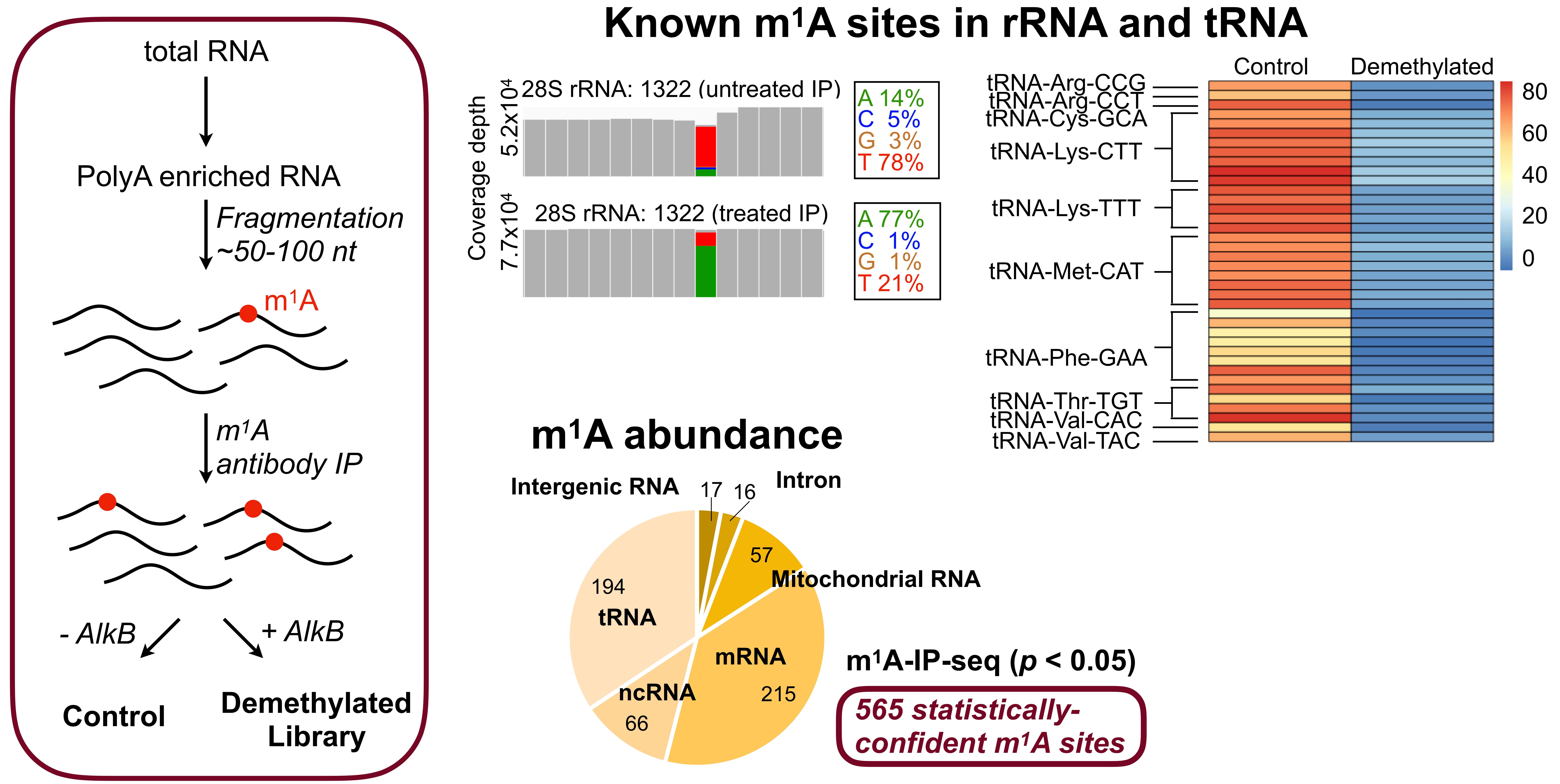
RT-1306 performed very well in in vitro assays, showing excellent product yields, mutational signatures, and breadth of sequence context around m1A sites. Critically, we could easily identify all of the known m1A sites on rRNAs and ribosomal RNA. More excitingly, because of this performance, we were able to perform unbiased analysis of m1A levels in human m1A samples - providing high-resolution maps of where m1A is present on mRNAs. Analyzing these datasets represented additional challenges - and due to some controversies with previous sequencing analysis methods, we developed fine-tuned, stringent bioinformatics pipelines to analyze our data.
Ultimately, our work led to a new method to map m1A in RNA samples. The key is that our new method is quite easy to use and should be deployable in other labs. Indeed, we have already shared our evolved enzyme with many other groups. More broadly, the selection platform we designed should be deployable for creating custom RTs for many other exciting and important RNA modifications. We are now working on scaling up our selection systems and creating new RTs for other RNA modifications. The key take-home message for us is that the marriage of directed evolution and synthetic biology to tackle problems in RNA regulation is a fruitful area for further research. Also, collaborating with friends always makes for fun science!
One final note is that Jane, the postdoc who led this work, did a phenomenal job. Bringing to bear protein engineering, molecular biology, sequencing, and bioinformatics - all at once - to develop the methodology and pipeline was truly an impressive undertaking. She is entering the academic job market this year, so if you are looking for an innovator in the RNA regulatory space - and someone to bring expertise in chemical biology and synthetic biology to your institution - Jane is truly an up-and-coming star.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in