MINK1 drives 5FU resistance via AKT/MDM2/p53 axis : Reveals a high throughput kinome knockout screening in OSCC.
Published in Cancer
The problem
Majority of head and neck cancer is originated from mucosal epithelium collectively termed as Oral squamous cell carcinomas (OSCC) (1). It is the most prevalent neoplasm in developing country like India with approximately 80000 new cases diagnosed every year (2). The treatment modalities for advanced OSCC include surgical removal of tumor followed by concomitant chemo-radiotherapy. Neoadjuvant chemotherapy is frequently prescribed for surgically unresectable OSCC tumor (3). However, despite of having all these treatment modalities, chemoresistance remains as one of the major causes of treatment failure in OSCC(4). The chemotherapeutic regimen used for OSCC are cisplatin, 5FU and Docetaxel (TPF)(3). Though chemotherapy drugs show initial positive response, tumor acquires resistance gradually and patients experience continued tumor growth and metastatic disease.
The purpose
Reprogramming resistant cells to undergo drug induced cell death is a feasible approach to overcome drug resistance. This can be achieved by identifying the causative factors for acquired chemoresistance and discovering novel agents to target critical causative factors, which will restore drug-induced cell death in chemoresistant OSCC. Previously, Kinases are known to play important role in regulating several phenotypes of carcinogenesis including growth, proliferation, angiogenesis, metastasis and evasion of antitumor immune responses [5]. However, studies relating kinase mediators of 5FU resistance in OSCC are limited. The goal of this study is to find out the potential kinase(s) those are major driver(s) of 5FU resistance in OSCC.
The approach
To understand the role of potential kinase signaling in 5FU resistance in OSCC, a CRISPR based kinome-wide high throughput screening was performed using a lentiviral sgRNA library knocking out 840 kinases individually with a total number of 3214 sgRNA constructs, followed by treatment with a sub lethal dose of 5FU for 48h after which cell death was measured in high content analyzer using a fluorescent cell viability dye. Further, to understand the kinase driven mechanism regulating 5FU resistance, we performed a phosphorylation profiling using a phospho-antibody array in presence or absence of the target kinase. In the end, to have a potential therapeutic approach, we looked for an efficient inhibitor against the target kinase in the international union of basic and clinical pharmacology (IUPHAR) database, where a screen of 72 inhibitors against 456 human kinases binding activity is provided.
.jpg)
The observations
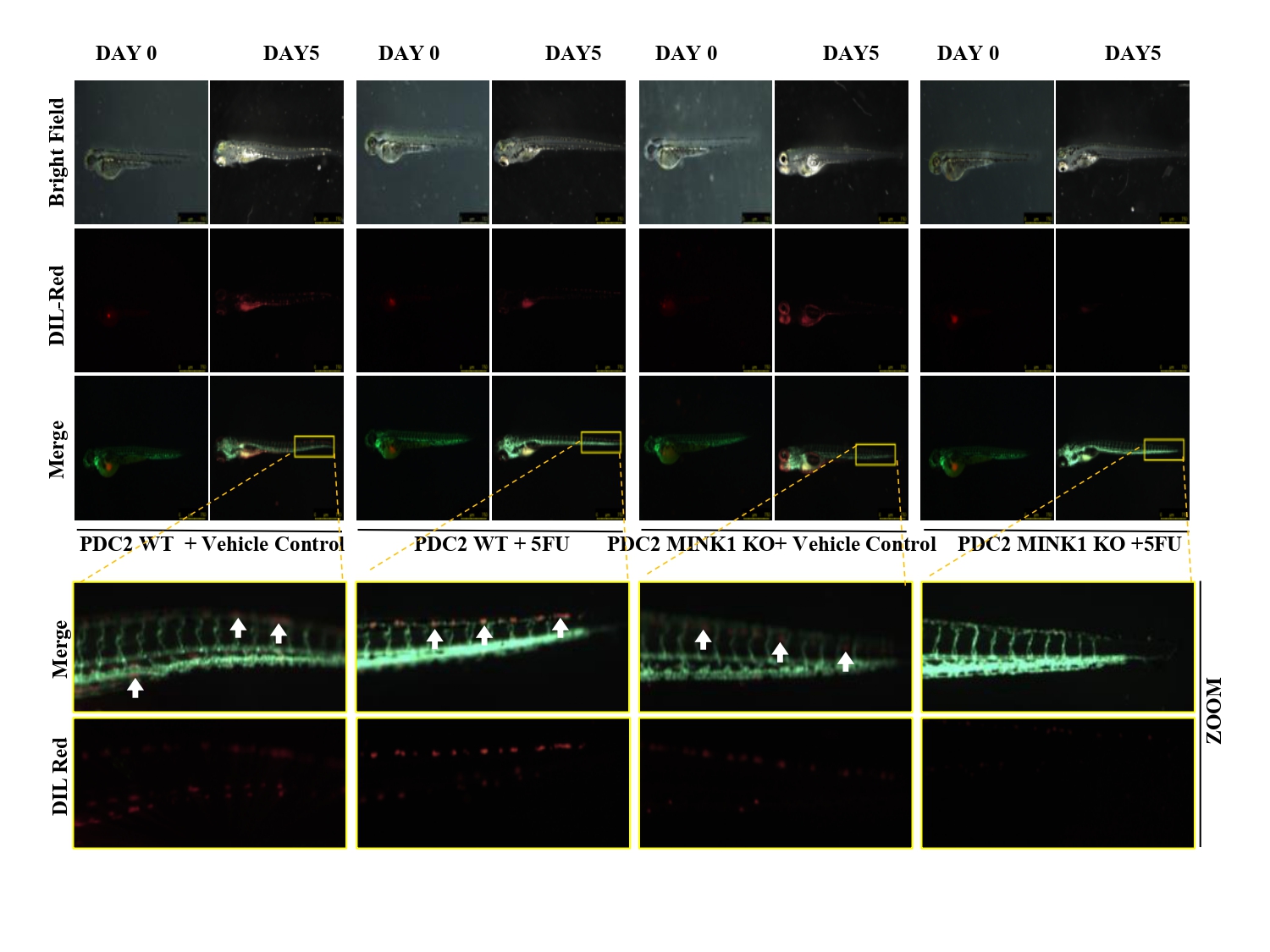
From the CRISPR/Cas9 based kinome knockout screening, performed in three different 5FU resistant lines developed in our laboratory conditions, MINK1 was found to be the top ranked kinase that can be targeted to re-sensitize drug resistant cells to 5FU (Fig. 1A-C). Analysis of clinical samples demonstrated significantly higher MINK1 expression in the tumor tissues of chemotherapy non-responder as compared to chemotherapy responders (Fig. 1D&E). The nude mice xenograft experiments indicate that knocking out MINK1 restores 5FU mediated cell death in chemoresistant OSCC (Fig. 1F). To confirm the finding from the kinome screening, cell viability and cell death assays were performed which suggest that knocking out MINK1 (MINK1 KO) significantly resenitized chemoresistant cells to 5FU (Fig. 2A&B).
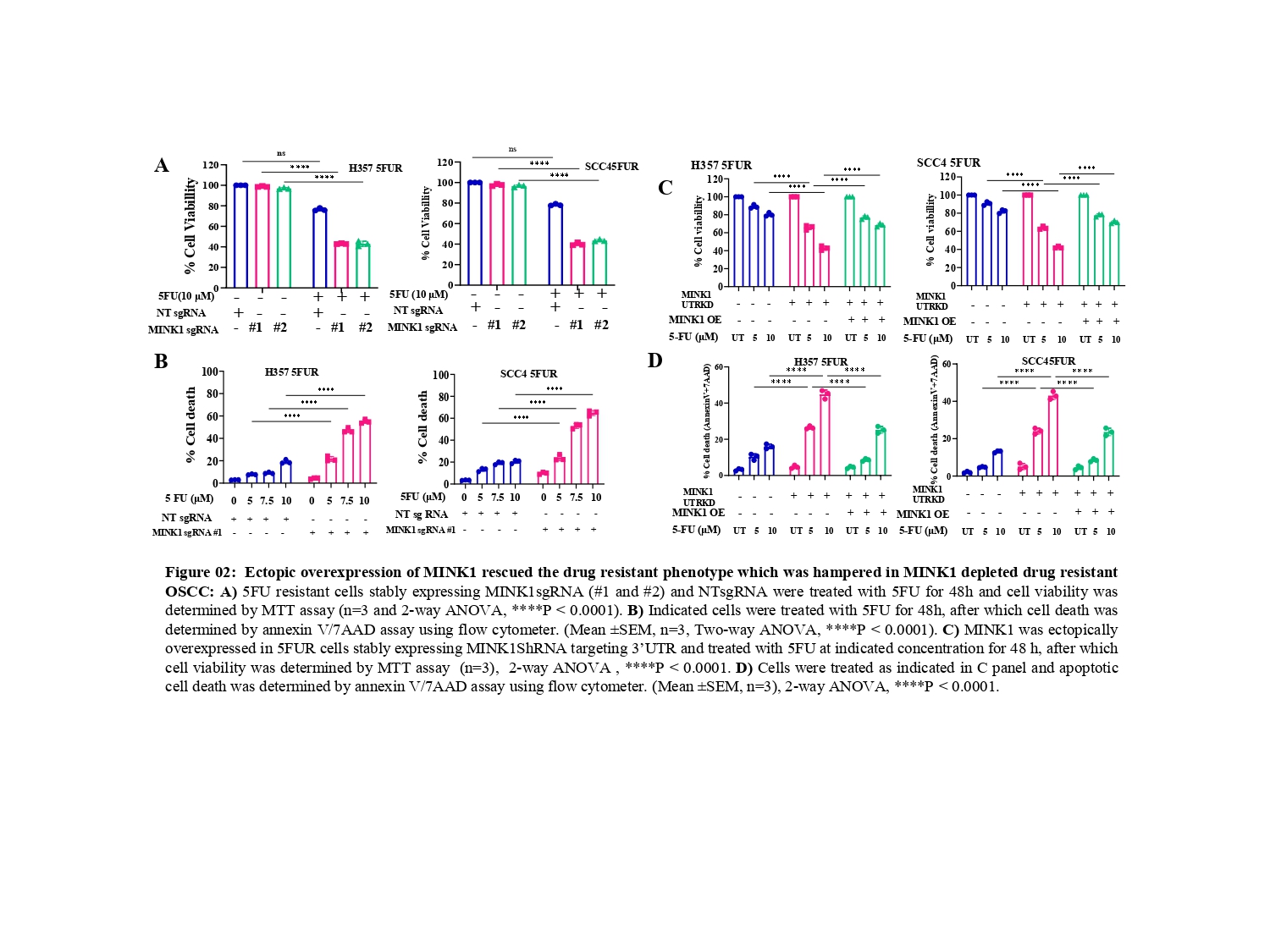
Next, Ectopic expression of MINK1 efficiently rescued the 5FU resistance phenotypes in MINK1 KD (knock down) OSCC (Fig. 2C&D), which further depicts MINK1 dependency of 5FU resistance in OSCC.
To understand the mechanism by which MINK1 regulates 5FU resistance, we performed a high-throughput phosphorylation profiling in MINK1 WT and MINK1 KO 5FU resistant cells (Fig. 3A&B). From this study, we found p- p53 (Ser33) and p-53(Ser15) to be significantly up-regulated in MINK1KO cells whereas p-AKT (Ser473) and p-MDM2 (Ser166) were found to be down-regulated in MINK1KO cells as compared to MINK1WT cells. These observations were further validated by immunoblotting assay. In line with these findings, we observed that ectopic overexpression of MINK1 reversed the expression pattern of all the above-mentioned genes (Fig. 3C&D). The tumor suppressor p53 is phosphorylated at various amino acids by different kinases, which tightly regulates its stability (5). It is well known that MDM2 (a E3 ubiquitin ligase) acts as a negative regulator of p53. MDM2 forms a complex with p53 and facilitates the recruitment of ubiquitin molecules for its degradation (6). Earlier, it was established that insulin induced activated AKT (Ser473) phosphorylates MDM2 at Ser 166 and Ser 186, which can lead to MDM2 mediated proteasomal degradation of p53 in cytoplasm as well as in nucleus (7, 8).
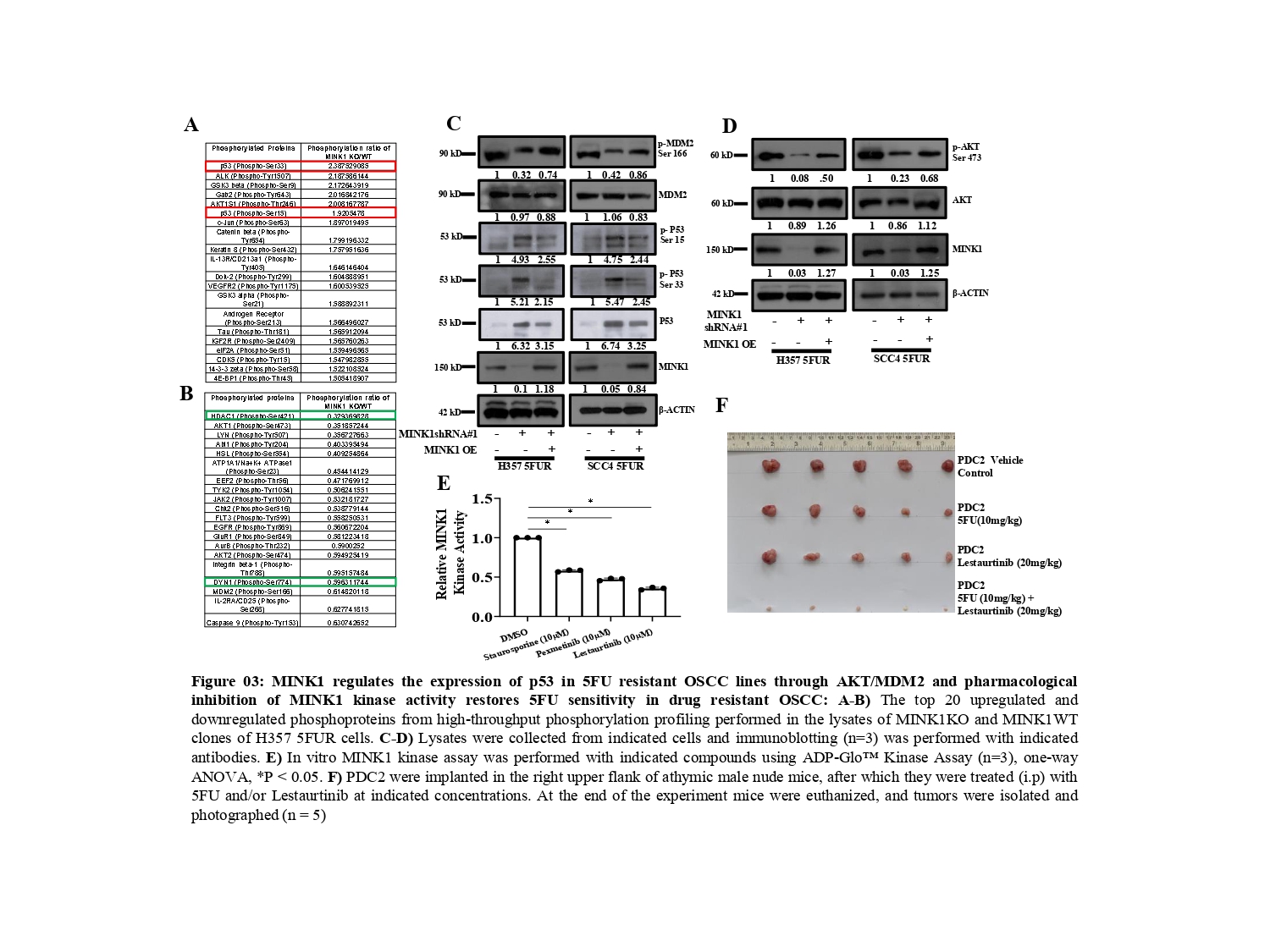
These events lead to blocking of p53-mediated transcription of genes those generally involve in apoptosis, cell cycle regulation and senescence. Phosphorylation of Ser15 and Ser33 leads to activation and stabilization of p53 as they attenuate the MDM2 mediated degradation of p53 (9, 10). Overall, in this study we found that MINK1 regulates the expression of p53 through activation of AKT which in turns triggers p-MDM2 (Ser 166).
Translational relevance
In search of developing a novel combinatorial therapeutic approach, we identified lestaurtinib as a potent inhibitor of MINK1 kinase activity among the top three small molecules predicted to bind to MINK1 as per the international union of basic and clinical pharmacology (IUPHAR) database (Fig. 3E). The patient derived TPF resistant cell-based xenograft data suggest that the novel combination of 5FU and lestaurtinib restores 5FU sensitivity and facilitates a significant reduction of tumor burden (Fig. 3F).
 Conclusions
Conclusions
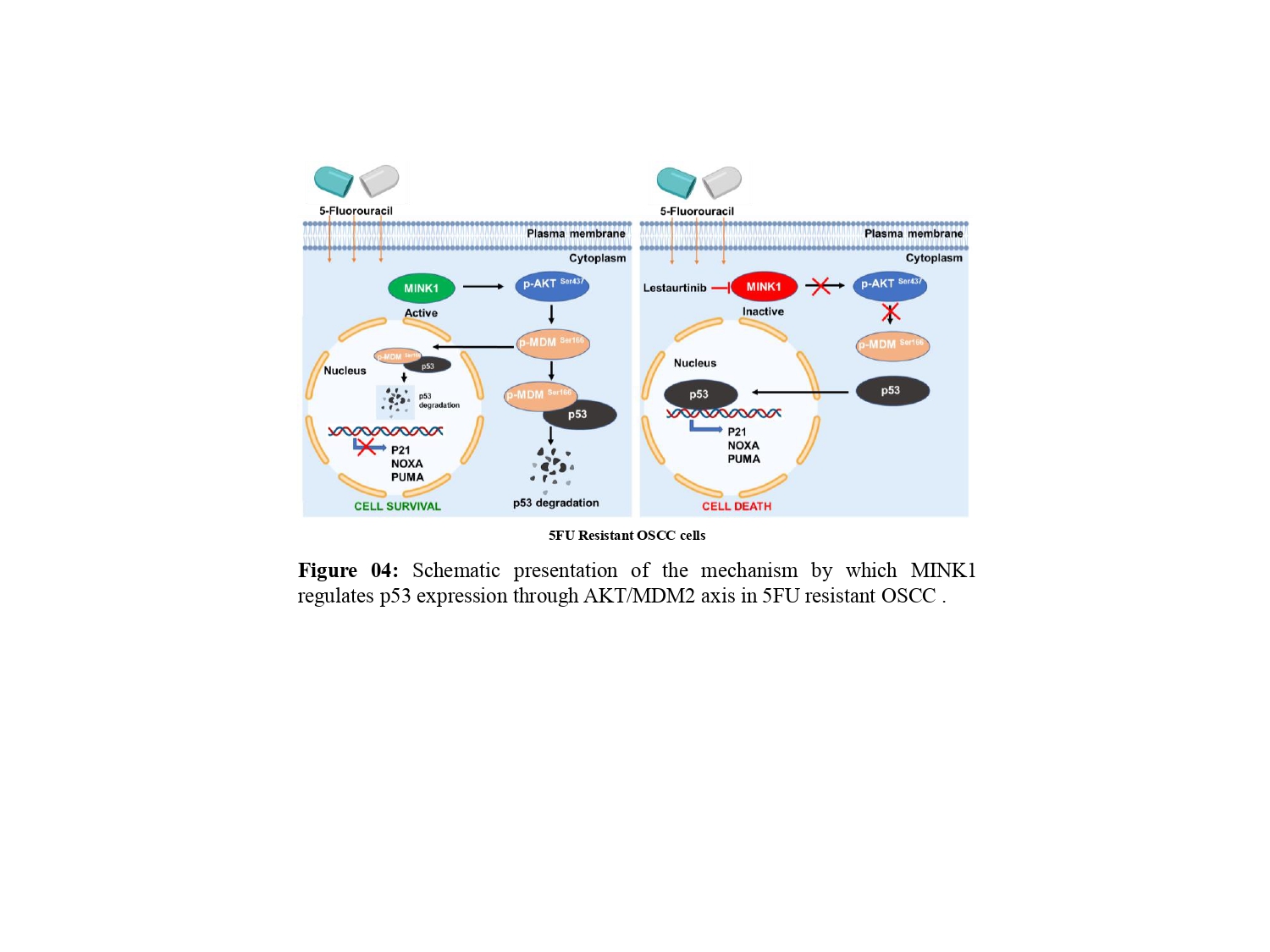
In this study, following the identification of MINK1 as a major driver of 5FU resistance, lestaurtinib was found to inhibit MINK1 kinase activity, which can reverse 5FU mediated cell death in chemoresistant OSCC lines. Ultimately, we demonstrated that MINK1 negatively regulates p53 in 5FU resistant OSCC by AKT-MDM2 axis, which in turn triggers degradation of p53 (Fig. 4).
Future avenues
Though we have demonstrated that MINK1 negatively regulates P53 through AKT/MDM2 axis in 5FU resistant OSCC, the 5FU specificity of this MINK1 driven signalling cascade remains to be fully elucidated through future studies. In addition, the novel combination of MINK1 inhibitor lestaurtinib and 5FU needs further clinical investigation in advanced OSCC.
References:
- Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. 2020. Head and neck squamous cell carcinoma. Nat Rev Dis Primers 6: 92
- Chaturvedi A, Husain N, Misra S, Kumar V, Gupta S, Akhtar N, Lakshmanan M, Garg S, Arora A, Jain K. 2020. Validation of the Brandwein Gensler Risk Model in Patients of Oral Cavity Squamous Cell Carcinoma in North India. Head Neck Pathol 14: 616-22
- Vishak S, Rangarajan B, Kekatpure VD. 2015. Neoadjuvant chemotherapy in oral cancers: Selecting the right patients. Indian J Med Paediatr Oncol 36: 148-53
- Maji S, Panda S, Samal SK, Shriwas O, Rath R, Pellecchia M, Emdad L, Das SK, Fisher PB, Dash R. 2018. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv Cancer Res 137: 37-75
- Maclaine NJ, Hupp TR. 2009. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging (Albany NY) 1: 490-502
- Moll UM, Petrenko O. 2003. The MDM2-p53 interaction. Mol Cancer Res 1: 1001-8
- Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. 2002. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 21: 1299-303
- Mayo LD, Donner DB. 2001. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A 98: 11598-603
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. 1998. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12: 2831-41
- Yogosawa S, Yoshida K. 2018. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci 109: 3376-82
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in