Mito-SiPE: Investigating low-frequency mitochondrial DNA mutations
Published in Protocols & Methods

Research examining the extent and impact of mitochondrial heteroplasmy is rapidly expanding. Mitochondrial DNA (mtDNA) mutations have been associated with a myriad of age-related and mitochondrial related diseases such as cancer, cardiomyopathy, and neurodegeneration1. These findings have put a spotlight on the mitochondrial genome. To investigate mitochondrial DNA mutations and further our understanding of these associations, it is important that researchers have a robust and sensitive method to detect and measure mtDNA mutations; which are referred to as heteroplasmy given the multi-copy nature of the mitochondrial genome. This usually requires separating out mitochondrial DNA from nuclear DNA. Most heteroplasmy studies to-date isolate mitochondrial DNA using long-range PCR amplification or probe hybridisation. These mitochondrial DNA enriched samples are sequenced and heteroplasmy is measured. These widely implemented methods have substantial limitations as described below.
Both methods rely on a sequence dependent method of enrichment i.e., specific primers or probes are designed to selectively enrich mitochondrial sequences from whole DNA extracts. This means they are susceptible to nuclear mitochondrial (NUMT) sequence contamination or co-enrichment2. NUMT sequences are segments or complete copies of the mitochondrial genome that have integrated into the nuclear genome over time. They are present in many species and the number and sequence of NUMTs is highly variable in humans3. Unfortunately, this means that selectively enriching only the DNA that resides inside a mitochondrion using primers or probes may not only be difficult, but perhaps impossible in certain circumstances. Unintentional capture of NUMT sequences is thought to explain the appearance of paternally inherited mitochondrial DNA reported in 20184.
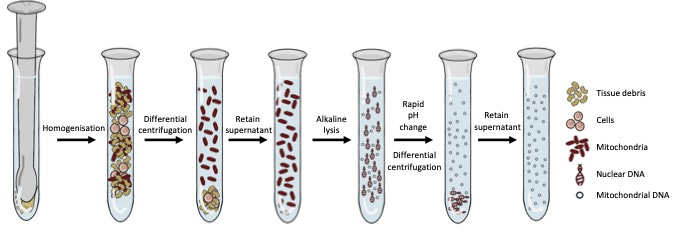
This challenge offered our lab the opportunity to adapt and characterise a method of mtDNA enrichment that was rooted in classic biochemistry to enrich mitochondrial DNA in a sequence-independent manner. This method relies on homogenisation, alkaline lysis, and differential centrifugation to first separate intact mitochondria (and the DNA inside them) from the rest of the DNA in the cell. This approach provides significant levels of mitochondrial DNA enrichment, without the use of sequence-specific probes or primers. Interestingly, when samples underwent enrichment with this methodology and were subsequently sequenced, we noticed that standard bioinformatic approaches for alignment and variant calling were suboptimal. We developed a bioinformatics pipeline to accompany the sequence-independent method of mitochondrial DNA enrichment and called this process Mito-SiPE (A Sequence-independent, PCR-free Enrichment of Mitochondrial DNA). Mito-SiPE was benchmarked against PCR amplification using Plog mutator mice to compare its performance in terms of heteroplasmy analysis. Plog mutator mice lack the proof-reading ability of their mitochondrial DNA polymerase and as a result, have elevated levels of mitochondrial DNA mutations compared to their wild-type counterparts5.
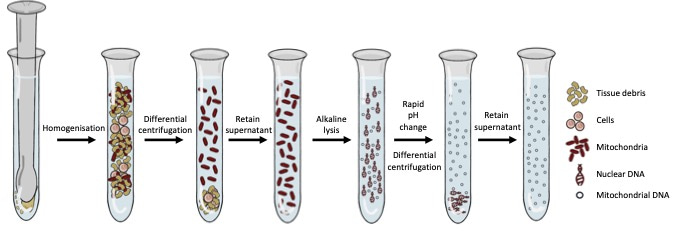 Figure 1: The wet-lab portion of Mito-SiPE which revolves around homogenisation, alkaline lysis, and differential centrifugation.
Figure 1: The wet-lab portion of Mito-SiPE which revolves around homogenisation, alkaline lysis, and differential centrifugation.
Heteroplasmy analysis of samples enriched using Mito-SiPE and PCR enrichment were comparable in samples that had high levels of mutation. However, Mito-SiPE avoided false heteroplasmic calls that were present in wild-type samples that had low levels of mitochondrial mutation when compared to PCR enrichment. Our findings suggest that PCR amplification leads to false heteroplasmic calls when investigating low frequency heteroplasmic variants. These results show that Mito-SiPE may offer researchers the ability to quantify low-frequency heteroplasmic variants that are unaffected by NUMT contamination or PCR errors.
The limitation of our methodology is that it requires a fresh and intact tissue to perform the initial homogenisation and alkaline lysis steps. Unfortunately, the current method is not compatible with previously purified existing DNA or small scale samples. Further optimisation of the protocol for samples with lower amounts of starting material might solve this issue. Additionally, the current limitations of low frequency mutation detection are restricted by the rate of sequencing errors produced by Illumina sequencing platforms. Our results demonstrate that we can consistently detect variation in the mitochondrial genome that occurs at a rate greater than 0.2%. Unique molecular identifiers (UMIs) have recently been used to decrease the sequencing error rate using consensus reads that are generated using the UMI barcodes. By combining Mito-SiPE with UMI technology, it may be possible to increase the sensitivity of Mito-SiPE. This is work that is currently ongoing and we aim to present our findings soon.
We hope that this methodology will be utilised by researchers who are interested in mtDNA mutations and will contribute to the rapidly growing and exciting field of mitochondrial DNA heteroplasmy and its role in human health and ageing
- Stewart, J. B. & Chinnery, P. F. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542 (2015).
- Parr, R. L. et al. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics 7, 185 (2006).
- Dayama, G., Emery, S. B., Kidd, J. M. & Mills, R. E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 42, 12640–12649 (2014).
- Balciuniene, J. & Balciunas, D. A Nuclear mtDNA Concatemer (Mega-NUMT) Could Mimic Paternal Inheritance of Mitochondrial Genome. Front. Genet. 10, (2019).
- Williams, S. L. et al. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 12, 675–682 (2010).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in