Modeling regulatory cis-elements for functional annotation of transcription-modulating genetic variants
Published in Protocols & Methods, Computational Sciences, and Genetics & Genomics

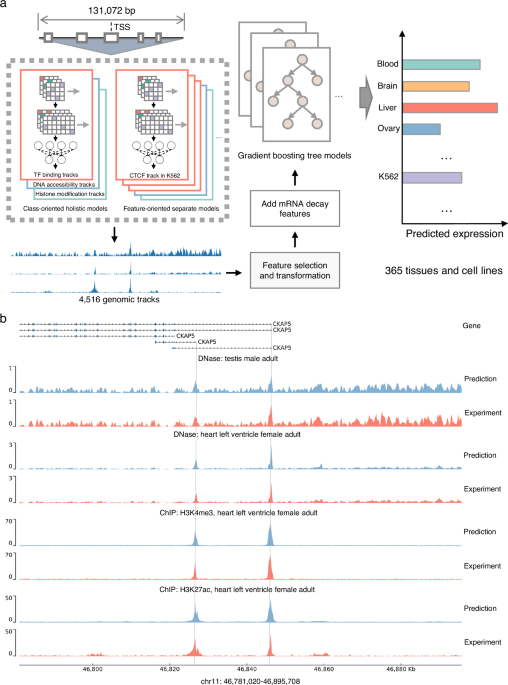
The human genome carries the instruction manuals for gene expression regulation. Deciphering the "genetic codes" encoded in human genome through computational approaches has emerged as a central theme in our lab.
Cells are the fundamental building blocks of life. Several kinds of functional cis- elements are involved in gene expression regulation of cells: more than 90% of the genetic variations linked to diseases and traits reside in noncoding regulatory regions. Modeling these noncoding cis-elements is one of key cornerstones for deciphering the cellular regulatory map.
Eukaryotic tissue-specific gene expression regulation relies on complex gene expression networks, involving the coordination of multiple regulators and specific DNA elements, which act in combinatorial manner. Inspired by our previous successful model design1, SVEN2 took a hybrid distinct model architecture other than the canonical "one-holistic-network-for-all" one: first learning "regulatory rules" from large-scale data through multiple class-oriented holistic models and feature-oriented separate models, and then applying these rules to infer tissue-specific gene expression level from sequences directly:
A set of sequence-based deep neural networks that learn regulatory codes from sequences to predict functional genomic features (TF binding, histone modification, and DNA accessibility). The basic idea here is to combine feature-oriented models (to learn the context-sensitive sequence-to-regulatory code, like the CTCF binding events in K562) and class-oriented models (to learn a more generalized rule, like TF bindings across different cell lines/tissues) for better utilizing available data.
A feature selection and transformation module to remove redundant features and reduce the dimensionality of the features.
A set of gradient-boosting tree models to predict gene transcription level based on transformed functional genomic features. Each model corresponding to one tissue or cell type.
SVEN’s modular design not only makes SVEN more interpretable, but also enables a customized model for users' particular tasks:

Benefiting from its unique design, SVEN shows consistently superior performance over canonical "one-holistic-network-for-all"-based Enformer in predicting tissue-specific gene expression level and assessing effects of variants on gene expression, with 40% smaller model size (153M for SVEN largest model and 249M for Enformer).
Noncoding variants that affect transcription are referred to as transcription-modulating variants. Several approaches have been developed to curate and characterize these variants effectively and efficiently, including our previous REVA database3 and CARMEN algorithm. Compared to small noncoding variants (≤ 50 bp), structural variants (SVs, > 50 bp) can have a more substantial impact on biological functions due to their larger scale.
The effects of SVs on gene expression were predicted by comparing the predicted expression levels of sequences containing reference alleles versus alternative alleles. In addition to its unique capability on quantifying transcriptomic impacts for large-scale SVs, SVEN can also infer tissue-specific gene expression profiles solely based on gene sequences. Notably, SVEN’s sequence-oriented design enables the identification of plausible underlying mechanisms for identified variants. The last but not the least, SVEN is also capable of handling small transcription-modulating variants.
We assessed SVEN's ability to predict the regulatory impact of SVs: SVEN demonstrated high accuracy, with a Spearman correlation of 0.921 between predicted and observed expression levels derived from paired RNA-seq data. Notably, the deletion upstream of the cancer biomarker PSMA-encoding gene FOLH1 disrupts the promoter region and the annotation-based algorithm predicted that this deletion would barely affect gene transcription; however, SVEN correctly predicted an increase in expression, partly because its annotation module indicated that the variant effectively increases expression-activating H3K4me3 and H3K27ac signals rather than the deleting known silencers or insulators. This finding suggests a plausible underlying mechanism for the observed effect of the deletion.

SVEN can accurately quantify the regulatory potential of genetic variants
There are still several paths for improving both accuracy and scalability. For instance, the three-dimensional structure of human genome mediates the interaction between regulatory regions and regulates gene expression. Incorporating predictions of the genome's three-dimensional structure could further enhance our approach. Fortunately, functional genomic data for the human genome continues to grow rapidly, allowing us to deepen our understanding of gene expression regulation on an ongoing basis.
Links:
- Full text: https://www.nature.com/articles/s41467-024-55392-7
- Source code of SVEN: https://github.com/gao-lab/SVEN
References:
- Shi, F. et al. Computational assessment of the expression-modulating potential for non-coding variants. Genomics, Proteomics & Bioinformatics 21, 662–673 (2023).
- Wang, Y., Liang, N. & Gao, G. Quantifying the regulatory potential of genetic variants via a hybrid sequence-oriented model with SVEN. Nat. Commun. 15, 10917 (2024).
- Wang, Y., Shi, F., Liang, Y. & Gao, G. REVA as a well-curated database for human expression-modulating variants. Genomics, Proteomics & Bioinformatics 19, 590–601 (2021).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in