Modeling solvent effects in chemical reactions
Most chemical reactions and nearly all biological processes occur in a liquid phase, with water being the most common solvent. Presence of the solvent molecules is crucial as they influence the stability of chemical species, the rate and mechanism of reactions, and the distribution of products. In organic chemistry, choosing the "right" solvent is key to the success of the synthesis. However, this choice is often based on empirical observations rather than a detailed understanding of how solvents influence reactions at the molecular level. While spectroscopic and computational methods are increasingly used to explore such effects, they often fall short of capturing the full complexity of these systems.
For us, computational chemists, balancing the accuracy and efficiency of our models is an everyday battle. The choice of the most suitable tools becomes particularly challenging for solvated systems, where the direct incorporation of solvent molecules leads to a significant increase in the system size. Modelling solvent effects can thus range from relatively cheap and simple continuum models, which approximate the solvent as a polarizable field, to computationally costly ab initio molecular dynamic (AIMD) approaches, where dynamical trajectories are generated using forces computed “on the fly” by solving the Schrödinger equation.
Frustrated by this ongoing struggle, our group turned to Machine Learning Potentials (MLPs) as an alternative to traditional classical and quantum methods for describing solvent effects. MLPs enable efficient mapping between nuclear configurations and energies/forces without the need to solve the Schrödinger equation directly for each structure. Moreover, unlike classical force fields, MLPs offer higher flexibility and the possibility for systematic improvement.
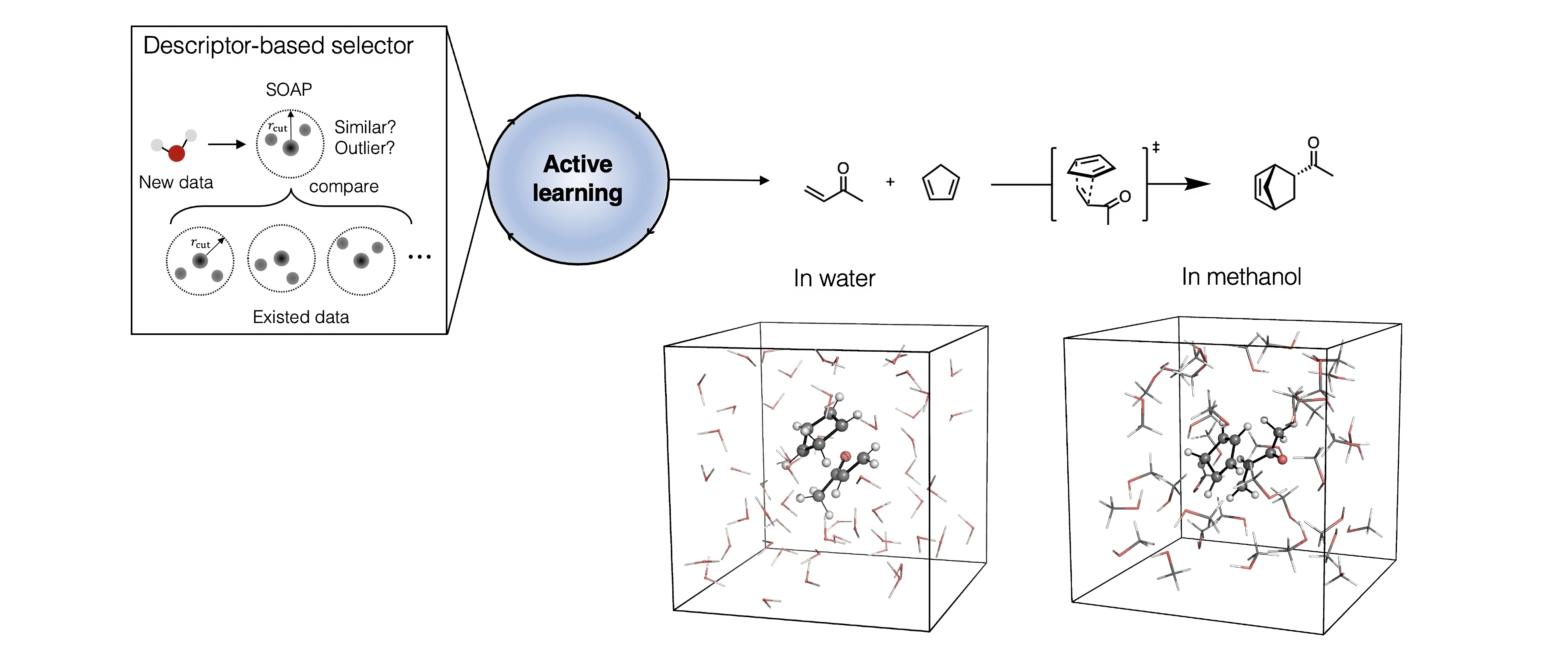
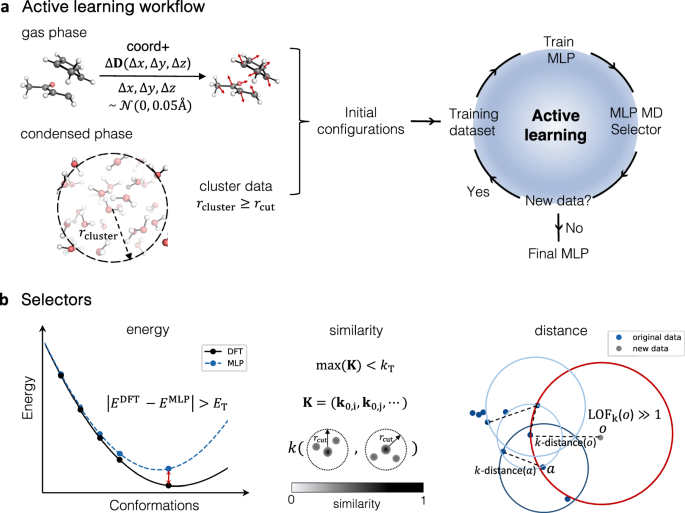
Building on previous work in our group, led by our colleagues Tom Young and Tristan Johnston-Wood, we implemented an Active Learning (AL) workflow to train reactive MLPs capable of describing organic reactions without relying on AIMD data. [1,2] As we began tackling more complex systems, we found that the bottleneck in the whole process was the selection of new and representative configurations to add to the training dataset. To address this, we focused on refining the structure selection process with two key improvements: defining a new selector and training on sub-systems that encompass intrinsic reactivity, and solute-solvent and solvent-solvent interactions. We used the Diels–Alder reaction of cyclopentadiene (CP) and methyl vinyl ketone (MVK) in explicit water and methanol as a representative system.
Traditionally, the selection step in AL strategies relies on variance in the prediction of energy and/or forces. The configurations with high variances are identified as under-represented and added to the training set. In our work, we used a slightly different strategy. Instead of looking into the variance, we investigate how the training data covers the potential energy surface (PES) represented in a feature space. To do so, we adopted the Smooth Overlap Atomic Positions (SOAP) descriptor to represent training data. [3] During the selection process, we either compare the SOAP similarity of the new configuration to existing data or determine if the new data point is an outlier to the training data set. We call this approach descriptor-based selectors.
We also introduced a computational strategy to build training data more efficiently by using knowledge of the specific chemistry being studied. Specifically, we combined data sets that represent the reaction under study with sets describing solvent-solvent and solvent-solute interactions. This approach, combining descriptor-based selectors and sub-system data sets, produces accurate and data-efficient MLPs using only 600 configurations, which contrasts with the several thousand required when using AIMD. The trained MLPs have already provided key insights into the origin of solvation effects on the Diels-Alder reaction, and will hopefully motivate the exploration of solvent effects more broadly. To facilitate this, we have automated the process and made it easy to use through our mlp-train package, which we continue to develop. We would welcome your feedback and suggestions.
[1] T. A. Young, T. Johnston-Wood, V. L. Deringer and F. Duarte, Chem. Sci., 2021, 12, 10944– 10955.
[2] T. A. Young, T. Johnston-Wood, H. Zhang and F. Duarte, Phys. Chem. Chem. Phys., 2022, 24, 20820–20827.
[3] A. P. Bart ́ok, R. Kondor and G. Cs ́anyi, Phys. Rev. B, 2013, 87, 184115.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in