More than just a toxin: Ammonia is a critical signaling molecule for activating lipid synthesis
Published in Cancer

The Center for Cancer Metabolism, a new research center at the Ohio State University James Comprehensive Cancer Center, has produced a major breakthrough in the scientific understanding of how tumor cells meet their abnormally high requirements for energy and cellular building materials by de-regulating lipid (fat and oil) synthesis.
This discovery from Deliang Guo, PhD’s lab group, spearheaded by research scientist Chunming Cheng, PhD, was the culmination of a ten-year effort to uncover how aggressive cancers such as glioblastoma (brain), breast, skin, lung, and many others are able to rapidly produce lipids used for creating new cells as well as for survival in low-nutrient environments such as the core of solid tumors, where access to energy sources like glucose (a basic carbohydrate, also known as blood sugar) and the oxygen to metabolize it are extremely limited.
In many incurable cancers such as the ones listed above, mutations in the genes that control EGFR (Epidermal Growth Factor Receptor) functioning lead to unchecked cell growth and replication. EGFR also tells the cell to take up abnormally large amounts of glucose from the blood stream. In cancers such as glioblastoma, chemotherapies that target EGFR fail because tumor cells have redundant mechanisms to EGFR that cannot be blocked. In these same cancers, lipid synthesis, or “lipogenesis” rates are extremely high compared to normal cells. Lipids such as fatty acids and cholesterol are the building blocks of new cells, forming the membranes around the cell itself as well as all its major and minor organelles like mitochondria. These cancer cells also create small organelles called “lipid droplets” that store excess fatty acids and cholesterols to be used when access to glucose and oxygen is limited by the location of the cell in the interior of the tumor.
The authors hypothesized that, if lipogenesis could be blocked, then this could open treatment options for incurable cancers with EGFR mutations by depriving cells of the lipids needed to grow and replicate. First, however, they needed to understand how lipogenesis was triggered in the first place. Cancer cells require massive amounts of energy and resources compared to normal cells. One way they get this is by taking up lots of glucose (influenced by EGFR) derived from dietary carbohydrates. The other method many cancers depend on is by taking up abnormal amounts of glutamine from the blood. Glutamine, an amino acid (building blocks of proteins), can be used by cells for energy via a process called glutaminolysis. Glutaminolysis splits glutamine into ammonia and glutamate. Glutamate is used in the mitochondria for cellular respiration, generating carbon dioxide and energy in a similar manner to glucose. Dr. Guo and Dr. Cheng hypothesized that cancer cells must be able to “sense” when there are excess energy sources available that can be used for lipogenesis, and so the “on-switch” must be related to cellular glucose and/or glutamine levels.
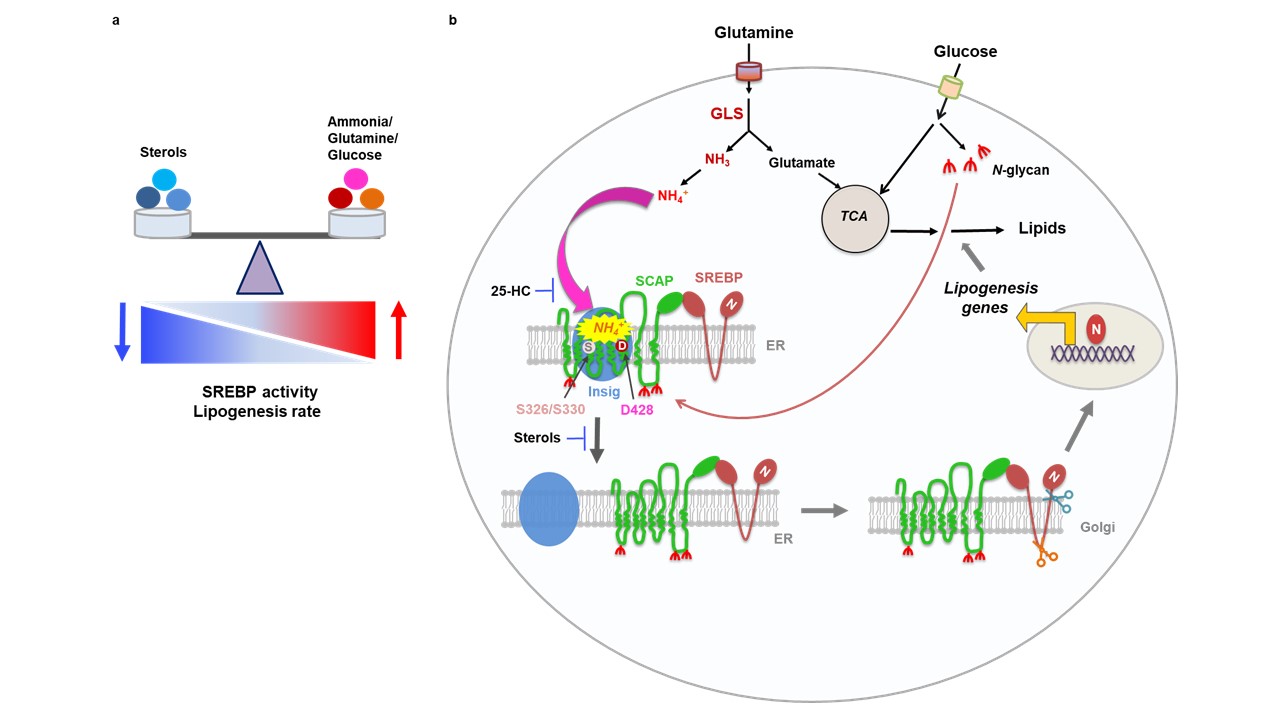
Lipogenesis is controlled by a regulatory protein called SREBP (Sterol-Regulated Element Binding Protein) that binds to DNA in the cell’s nucleus and activates lipogenesis genes. SREBP is formed in the endoplasmic reticulum (or ER – the cellular organelle that manufactures both proteins and lipids), where it binds to another protein called SCAP (SREBP-Cleavage Activating Protein). SCAP binds to another protein called Insig (INSulin-Induced Gene). Insig’s bond to the SCAP/SREBP complex prevents it from leaving the ER unless the cell “senses” that conditions are right for lipid synthesis. When it does, SCAP releases from Insig and transports SREBP to another organelle called the Golgi Apparatus, where two special enzymes cleave SREBP into fragments. One of these fragments, the N-terminus, can enter the nucleus, bind to DNA and activate lipogenesis.
Previous research showed that sterols including cholesterol and 25-hydroxycholesterol (25-HC) can bind to SCAP in the ER. When it does, SCAP changes shape and binds Insig tightly to SCAP/SREBP, preventing SREBP from leaving the ER and stopping lipogenesis. This prevents the cell from making too many lipids, which can build up to toxic levels and kill the cell (acting as an “off switch” to lipogenesis). Cancer cells can evade this shutoff mechanism by storing lipids like cholesterol in lipid droplets to continuously synthesize new lipids.
Dr. Cheng’s early experiments revealed something unexpected: both glucose and glutamine were required by cancer cells to activate the genes controlling lipogenesis, even when sterols were removed from cells. It made sense that glucose must be involved, as it is the most basic source of metabolic energy for cells and also a building block for lipids. The fact that lipogenesis could not occur without glutamine despite the cells being provided with excess glucose was very surprising. Conversely, when provided with glutamine alone, lipogenesis could not occur. Dr. Cheng had uncovered evidence of the cellular trigger for lipogenesis. The question was, how exactly did it work?
After five years of comprehensive experiments, Dr. Guo’s team found the first answer: SCAP “senses” glucose levels in the cell. When glutamine was provided alone, SREBP stayed bound in the ER, but interestingly, SCAP disappeared entirely. It seemed that SCAP was unstable and would degrade without glucose. Without SCAP, SREBP could not be transported to the Golgi Apparatus. They found that a process called N-glycosylation modification, where glucose is converted to the substrate N-glycan, stabilizes SCAP. N-glycan links to SCAP in three places and prevents it from degrading. This meant that cells could “sense” when glucose was available for lipogenesis because, without stabilizing SCAP, it was impossible to activate SREBP (Cheng et al. 2015, Cancer Cell). What was still missing however was the trigger that caused SCAP to unbind from Insig.
Five years later, at the culmination of a decade of research with Dr. Guo’s research team, Dr. Cheng found something amazing. He knew that glutamine was necessary to trigger lipogenesis genes. However, he found it was not glutamine itself that was the trigger – it was ammonia from glutaminolysis that could trigger SCAP to unbind from Insig. Ammonia, a small molecule made of one nitrogen atom and three hydrogens, was historically thought to be nothing more than a waste molecule. It is a toxic byproduct of protein metabolism that is filtered out of the blood and detoxified as urea in urine. Recent research showed that some cancer cells could recycle it to make more amino acids (to build more proteins), but never before had someone shown that ammonia could be a biochemically significant signaling molecule. Computer models of SCAP’s structure identified three binding sites for ammonia that cause SCAP to change shape and let go of Insig. Amazingly, these sites were very close to where 25-HC binds to SCAP. In normal cells, 25-HC could block ammonia from SCAP, preventing lipogenesis when cholesterol levels are high in the cell. In cancer cells that create lipid droplets, ammonia from abnormally high glutaminolysis rates combined with abnormally high glucose/N-glycan levels result in a deadly combination that allows the cell to create lipids unchecked, providing for a future of indefinite growth and replication.
The Guo lab’s incredible discovery links together cellular metabolism of the three major classes of dietary macromolecules: fats (lipids), amino acids (glutamine specifically) and carbohydrates (glucose). This discovery also opens the door to a treatment revolution not just for untreated cancers and potentially other metabolic diseases as well. Dr. Guo’s lab has already shown that, by blocking SCAP activation by ammonia, lipogenesis can be shut down, leading to the starvation, shrinkage, and elimination of tumors regardless of EGFR signal de-regulation. These results offer great hope for the development of treatments for untreatable cancers like glioblastoma.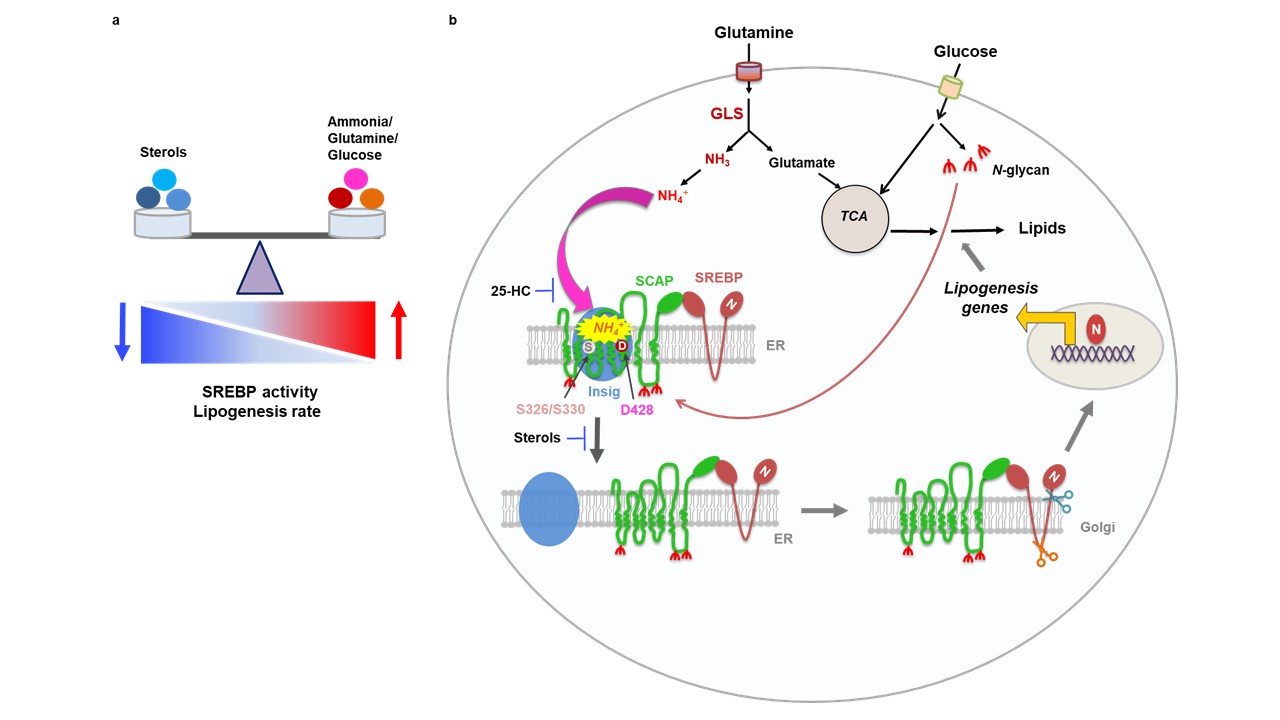
Figure: figure that shows the lipogenesis biochemical pathway. Part a describes SREBP activity. When sterols increase, 25-HC binds to SCAP, reducing SREBP activity and reducing lipogenesis. When sterols are low and glutamine and glucose are high, ammonia causes SCAP to dissociate from the ER, activating SREBP. Formation of lipid droplets in cancer cells prevents sterols from competing with ammonia for SCAP activation sites.
This amazing discovery was published online by the journal Nature Metabolism (Cheng et al., 2022) and can be found at the following link: https://www.nature.com/articles/s42255-022-00568-y
To learn more about the Center for Cancer Metabolism and the groundbreaking research by its members, please visit us at our website: https://cancer.osu.edu/cancermetabolism
Follow the Topic
-
Nature Metabolism
This journal publishes work from across all fields of metabolism research that significantly advances our understanding of metabolic and homeostatic processes in a cellular or broader physiological context, from fundamental cell biology to basic biomedical and translational research.

Related Collections
With Collections, you can get published faster and increase your visibility.
The expanding therapeutic landscape of GLP 1 receptor agonists
Publishing Model: Hybrid
Deadline: Jan 23, 2027
Microbiome and energy metabolism
Publishing Model: Hybrid
Deadline: Dec 06, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in