Multiplexed phosphoproteomics of low cell numbers
Published in Protocols & Methods and Cell & Molecular Biology

How does a cell decide whether to divide, differentiate, migrate or die? Understanding cellular decision-making relies on measuring the biochemical signals regulating these processes. These signals are transmitted via phosphorylation - the reversible attachment of a phosphate modification that acts as a molecular switch to regulate proteins. Unlike protein sequences, these modifications are not encoded in our genome, instead, they are controlled in a stimulus-responsive manner by enzymes - kinases and phosphatases. Mass spectrometry is the sole unbiased technology capable of quantifying these transient phosphorylation events across the proteome (phosphoproteomics). Owing to the stoichiometric nature of phosphorylation, an additional processing step is required to enrich phosphorylated peptides which poses a significant challenge when dealing with limited material. As we move beyond bulk‑average measurements, the study of rare cell populations is becoming increasingly important in the context of developmental subtypes, treatment response, origins of disease, and primary patient material. While genomics and proteomics enable the routine analysis of single-cells, phosphoproteomics, has lagged behind. The gap between what we want to measure and what we can measure has been widening. Our goal was to develop a method that could quantify phosphorylation site changes from just 1,000 cells without compromising on depth, reproducibility, or practicality. Here, we introduce SPARCE (Streamlined Phosphoproteomic Analysis of Rare CElls), outline how it works, what we learned while developing it and why we think it opens new doors for understanding signalling in rare cells.
Less is more
Fluorescence‑Activated Cell Sorting (FACS) is the workhorse for isolating small cell populations of interest, but it leaves cells in a saline buffer, which interferes with downstream proteomic workflows. Integrating FACS into proteomic-compatible sample preparation techniques has historically been overlooked. Conventional workflows use harsh lysis buffers followed by multiple processing steps — dilution, pH adjustment, desalting — all of which reduce yield and reproducibility when applied to low-input samples.
SPARCE was designed to minimise sample loss and streamline the processing of FACS-sorted cells by combining four core elements (Figure 1):
- Water-based lysis. Using water, freeze-heat cycles and sonication to release proteins directly from cells eliminates the need for detergents or chaotropic agents. This not only simplified the workflow but also improved digestion efficiency - we saw a fivefold increase in fully digested peptides compared to urea lysis.
- Single-step digestion. We found that including protease and phosphatase inhibitors reduced digestion efficiency. The same was true for reduction and alkylation, possibly because the reducing agent destabilises trypsin itself. So we omitted both and increased the trypsin-to-protein ratio, which significantly enhanced the number of peptides identified.
- On-tip TMT labelling. This allows peptides to be simultaneously desalted and labelled on a C18 tip, improving labelling efficiency and peptide retention. This approach reduced sample handling, increased the yield of hydrophilic peptides, and almost doubled identification rates relative to in-solution labelling.
- Multiplexed phosphopeptide enrichment. Multiplexing of labelled samples before phosphoenrichment enables reproducible enrichment across conditions. Multiplexing also facilitates the addition of a carrier channel to boost total protein amount and signal intensity without introducing measurable ratio compression when acquisition parameters are optimised.
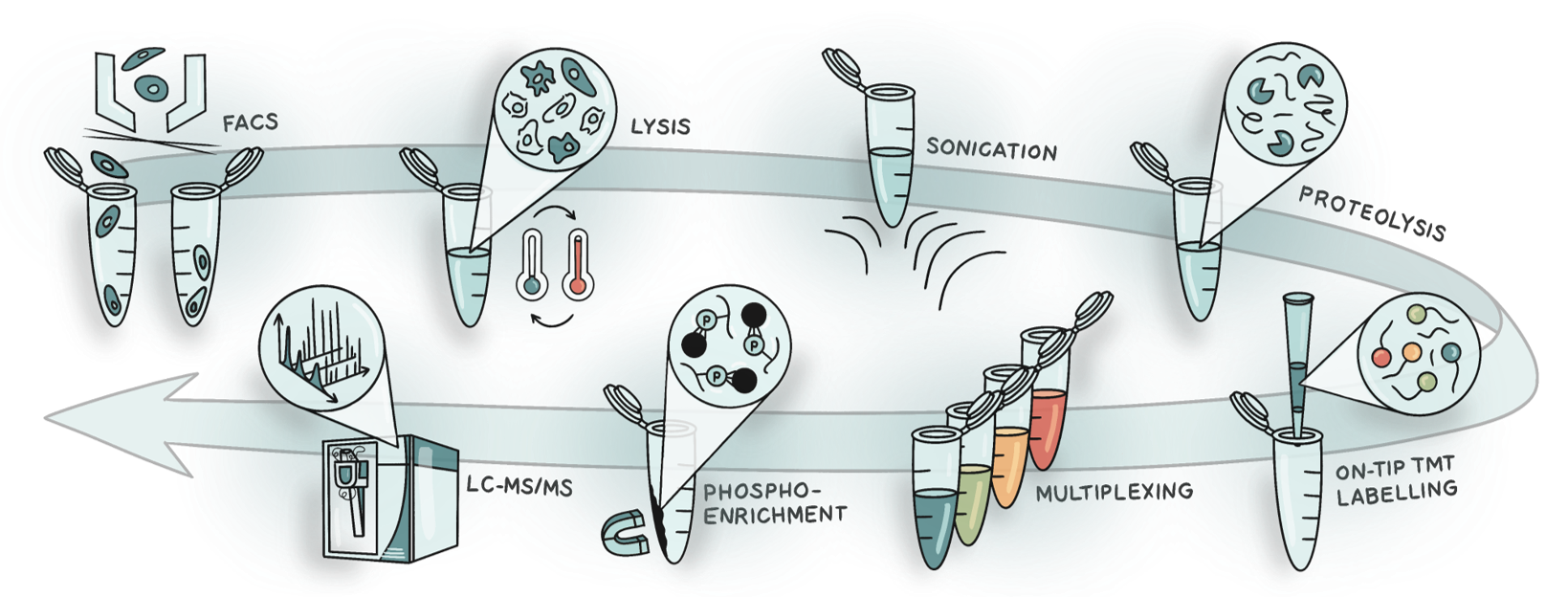
Figure 1. Schematic of the SPARCE workflow. Cells are isolated using FACS and subjected to lysis by freeze-heat cycles and sonication. Proteins are digested, and peptides are labelled and desalted on the same tip. Labelled samples are multiplexed, and phosphopeptides are enriched for LC-MS/MS analysis.
Putting SPARCE to the test
To test whether SPARCE could detect biologically meaningful and reproducible signalling changes across different samples, we analysed four patient-derived glioblastoma stem cell lines. Each was profiled using both 20,000 (high-input plex) and 1,000 (low-input plex) cells, the latter with an additional carrier channel containing a mixed pool of cells from the different samples. Almost 3,000 well-localised phosphorylated peptides were identified in the low-input plex, and ~4,500 in the high-input plex. Principal component analysis and hierarchical clustering showed that biological variation between cell lines outweighed technical variation arising from differences in cell input levels. Kinase‑substrate enrichment highlighted differences across cell lines, reflecting underlying variation in cell cycle and stress response pathways, which may be relevant for understanding tumour heterogeneity and therapeutic resistance. Importantly, functional analysis revealed that the distinct kinase activity patterns between cell lines were reproducible across both the high-input and low-input plexes, confirming that SPARCE can faithfully capture biological changes when material is scarce.
What we’ve learned
In developing SPARCE, we found that many assumptions in proteomics workflows don’t hold true for processing nanogram-scale samples. For instance:
- Protease and phosphatase inhibitors may not be necessary, and in fact, may interfere with digestion.
- Reduction and alkylation can reduce yield when working with nanogram amounts of protein.
- Trypsin needs to be in excess, far beyond what’s used in high-input protocols, to ensure complete digestion.
- On-tip labelling improves yield and efficiency, especially for hydrophilic peptides, and streamlines sample processing compared to in-solution labelling.
Looking forward
SPARCE offers a practical solution for phosphoproteomic profiling of rare cells. By simplifying lysis, minimising sample transfer steps, and integrating labelling with desalting, it avoids many of the limitations associated with traditional methods. Its compatibility with FACS makes it especially well-suited for biological studies involving tumour subtypes, immune populations, or developmental phenotypes.
We hope SPARCE will be useful to others who are trying to study cell signalling in rare populations. It doesn’t require specialised instrumentation or expensive reagents and can be used with conventional TMT kits, standard LC-MS systems, and existing FACS setups. We’re continuing to test SPARCE in other contexts to expand its utility, but for now, we think SPARCE offers a simple, reliable, and scalable way to quantify signalling changes in small samples. In a field where sample availability often determines the scope of the question, SPARCE offers an opportunity to ask new ones.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in