Nominal data mining and precision medicine
Published in Cancer

Imagine yourself sitting on wooden platforms during a sunny and humid day, watching a medieval knights tournament at Visegrád Castle, when a light on "multipartite networks" flashes across your head. This performance was part of the interdisciplinary signalling workshop (ISW2017) in Hungary in July 2017, which did not finish with sitting and watching. The audience (conference participants) were also becoming medieval knights, competing in combat skills in groups of ten with spears, swords, and other weapons. It was an opportunity among other memorable networking possibilities at this conference, for me not only to present my idea to other fellow scientist friends and colleagues, but also have time to relax, have a few laughs together, and get enthusiastic about challenging training goals (due to the restrictions of the coronavirus pandemic, I feel even more the necessity for similar conferences with networking events). My first memory of conceiving the idea was here, and it gradually developed until I moved to the Institute for Molecular Medicine Finland (FIMM) at the University of Helsinki in Finland as a senior researcher almost a year later, in October 2018. Although I was surprised at being told upon arrival to prepare a grant proposal, I quickly realised that, thanks to a collaboration with Caroline Heckman, I had access to datasets at FIMM1 with the exact attributes I required to implement my idea, in particular, datasets containing multi-level nominal variables based on patients. It was at this point that I felt it was the right time and the right place to focus on a cancer disease like acute myeloid leukaemia (AML), which has a poor prognosis and requires new treatment options and drug combinations. This article is the first piece of work, which was done as a pilot study on a database comparable to the one we used2.
 |
 |
 |
 |
 |
 |
A few scenes from the knights' tournaments and medieval team-building activities of researchers on the sidelines of the ISW2017 conference
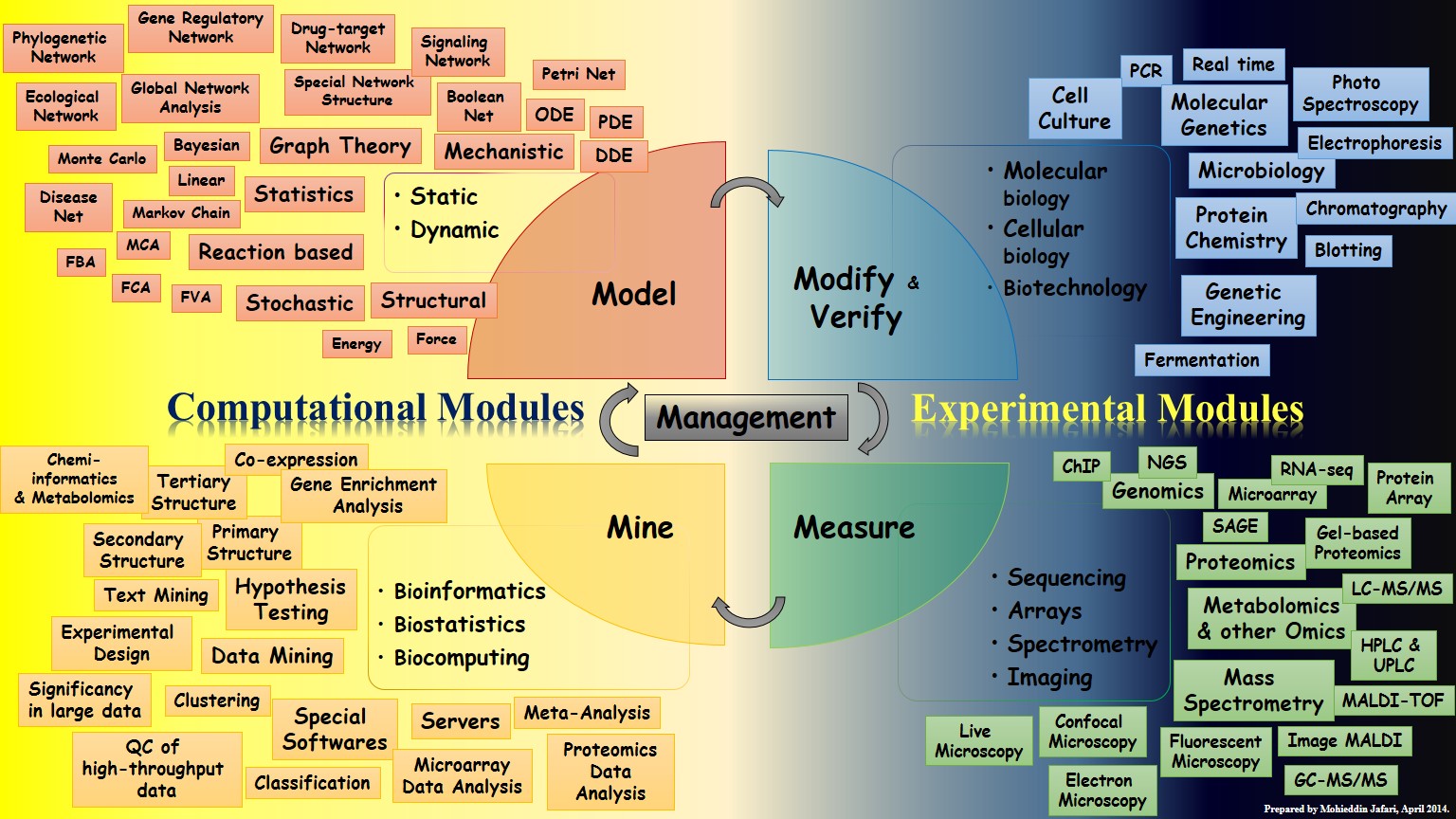
The study design was based on a systems biology paradigm, as indicated in Figure 1 of the article. That is the modify-measure-mine-model paradigm3, which has been accompanied by prediction verification. After preprocessing, drug response datasets that contain a significant number of patients and drugs were used to construct bipartite networks, and preparations were made to recommend beneficial therapeutic combinations by clustering patients and drugs. Other supporting evidence for the validity of the predictions was assessed using prior knowledge, and ultimately, more experimental evidence was acquired in the lab.
 |
|
.jpg)
The paradigm of systems biology and the study's flowchart
As a result, this can be considered a typical systems biology study since it includes thorough computational and experimental modules, consistent with a holistic view of systems biomedicine that incorporates interconnections between entities. The key to achieving acceptable and relevant results in such studies is to use a common language and to communicate effectively across researchers from distinct disciplines. Since this kind of research usually starts with a problem in biomedical topics and continues with solutions from other fields like computer science, mathematics, and statistics. I don't think the necessary interaction will be achieved if researchers with diverse backgrounds look at each portion as a black box, focused just on the result of each section, and are relatively unaware of the details of the analysis. Having people with an interdisciplinary background in a team to build effective communication has always shown to be a winning formula. For example, as shown in Figure 2 in the article, before using the common measures for drug response, namely AUC and IC50, the possibility of using them was examined in order to better compare the drug responses. Given the problems that these two criteria have, as described in the article, we finally developed the model by choosing the median of cell viability.
The significance of this work can be summarised from a theoretical point of view to a therapeutical perspective. The primary goal of the study from the outset was to gather additional information from patients based on the treatment response of ex vivo samples in order to recommend their optimal drug combinations. In other words, because drug response involves information at the cellular phenotypic level, we assumed that it could better reflect cell signalling events in the laboratory and, subsequently, in the clinic4. However, while achieving positive results on patient clustering and medications, confirmed by independent evidence, the nature of the data drew my attention once more. I am referring to the nominal nature of the data that I used to build and analyse the model. I realised I was doing nominal data mining, and this method could be used to extract information from other datasets with at least two nominal variables with several levels. On the other hand, some of the similarity measures computed for drugs and also for patients in this study are noteworthy and could be the subject of further research. Consider Figure 5 in this article, which provides independent evidence to support cluster homogeneity analysis. We believe that drug similarity in terms of SMILE structure (Simplified Molecular Input Line Entry System) or target proteins, as well as their clustering into two distinct clusters, as opposed to random grouping, is remarkable. Note that our findings are based on the drug sensitivity analysis of a single type of disease, i.e., AML, and these drugs are thought to be functionally similar (mostly kinase inhibitors). Furthermore, the strategy used in this study to propose drug combinations is therapeutically promising in instances where there is an exponentially different number of states for drug combinations. Considering patient heterogeneity, our approach would recommend various combinations that can be successful in the majority of patient samples (not shown in the linked article). Adding a few tests based on the proposed drug combinations is not much of an operational hassle, especially in circumstances where automatic liquid handling systems use technology to test a large number of drugs in a short period of time.
We would be thrilled if you enjoyed reading this paper and decided to apply the suggested approach to similar datasets since our team has also developed an R package called NIMAA5 for this purpose. Finally, I would like to convey my appreciation to all of my coworkers and friends (Mehdi Mirzaie, Jie Bao, Farnaz Barneh, Shuyu Zheng, Johanna Eriksson, Caroline Heckman & Jing Tang) who assisted me in completing this article. Aside from that, I would like to express my admiration for the collaborative and amicable attitude that exists inside the Systems Oncology Research Unit (ONCOSYS), which enables this research to be conducted.
.jpg) |
.jpg) |
Network pharmacology for precision medicine group at Helsinki University
References:
1. Malani, D. et al. Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer Discovery, candisc.0410.2021, doi:10.1158/2159-8290.CD-21-0410 (2021).
2. Tyner, J.W., Tognon, C.E., Bottomly, D. et al. Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531 (2018).
3. Aldridge, Bree B., John M. Burke, Douglas A. Lauffenburger and Peter K. Sorger. “Physicochemical modelling of cell signalling pathways.” Nature Cell Biology 8 (2006): 1195-1203.
4. Yaffe MB. Why geneticists stole cancer research even though cancer is primarily a signaling disease. Sci Signal. 2019 Jan 22;12(565):eaaw3483.
5. Jafari, M., Chen, C., Mirzaie, M. & Tang, J. NIMAA: an R/CRAN package to accomplish NomInal data Mining AnAlysis. bioRxiv, 2022.2001.2013.475835, (2022).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in