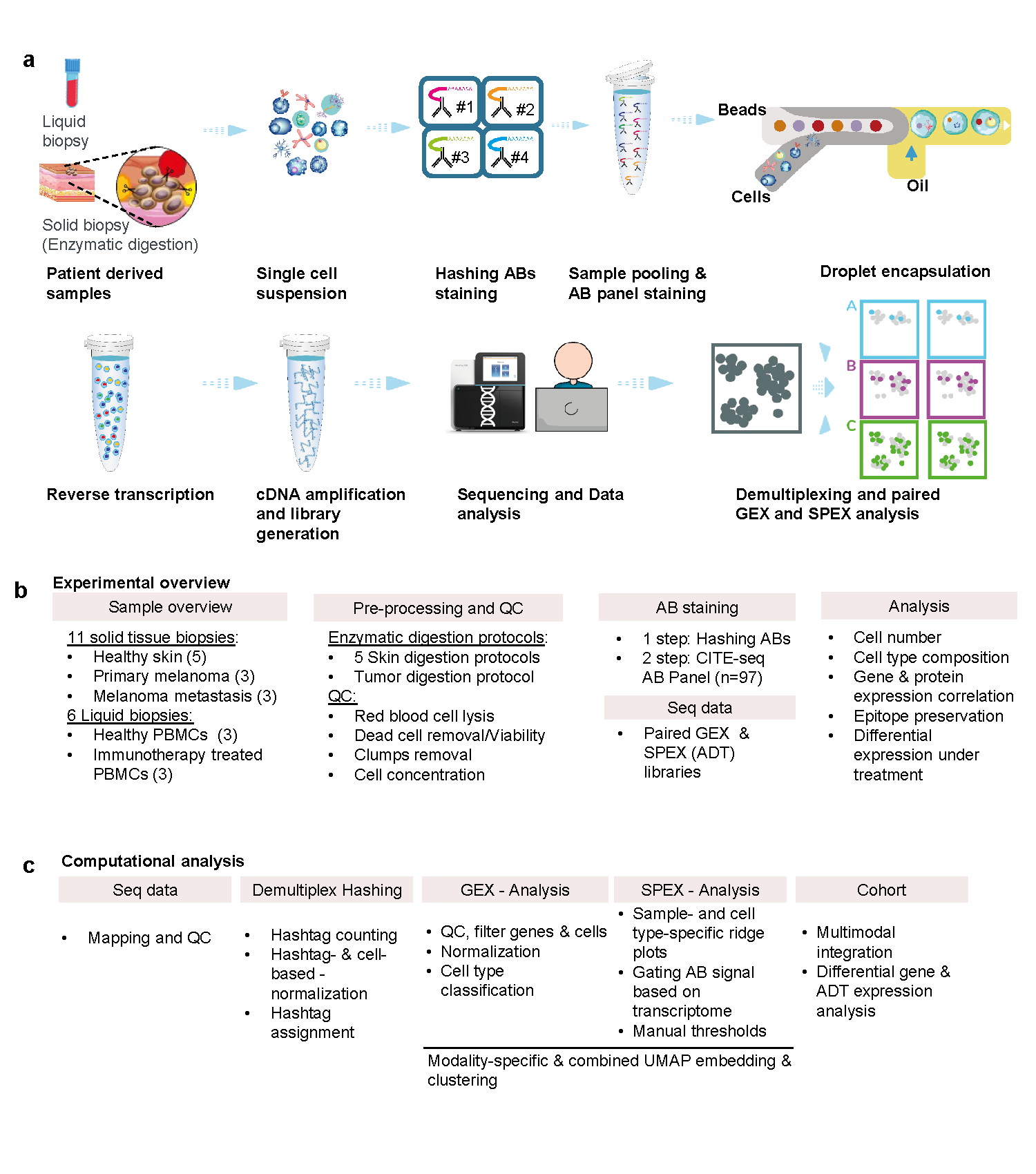

Single-cell profiling advanced our understanding of the molecular complexity of cancer. Individual omics methods, particularly single-cell RNA sequencing has revolutionized the resolution we can look at the heterogeneity of cancer and its microenvironment. However, in many cases, both tumors, as well as immune infiltrates, are only profiled on the transcriptomic level, which limits our understanding of the complex tumor multi-layered molecular landscape and overlooks changes beyond RNA expression.
In the current study, we focused on several aspects of Cellular Indexing of Epitopes and Transcriptome by Sequencing (CITE-seq) multi-omics profiling that allows quantitative targeted profiling of surface proteome and global transcriptome at a single-cell level1. The technology utilizes oligo-labeled antibodies (ABs), where antibody-derived tags (ADT) identify unique protein counts and the number of ABs that can be added to the panel is practically unlimited and exceeds the number of available antibodies today. The additional advantage is the possibility of adjusting antibody signal by “gating” on transcriptome-typed cell populations and applying dynamic thresholding of the signal. This allowed us to skip the usually required antibody titration step, which is impossible for large panels of ABs due to cost and instead performed the computational adjustment to discriminate between the positive and negative signals.
CITE-seq is a promising multi-omics technology, that can be optimized to study clinical samples. To achieve this, first, we looked into how pre-processing of patient-derived samples can bias the results. Particularly enzymatic dissociation necessary to obtain a single-cell suspension compromises the surface epitope expression. Differential gene expression of enzymatically treated and untreated immune cells identified activation of innate immune, stress as well as heat shock response pathways. Additionally, by performing differential protein abundance and applying the dynamic thresholding we observed not only loss of surface epitopes on all cell populations but also artifactual gain of surface markers associated with antigen presentation, cell adhesion, and migration processes. This suggests that several hours of pre-processing was enough to cause in vitro immune cell activation, which might be misinterpreted as an in vivo biological process.
Second, we investigated the overlap in the co-detection of RNA and the corresponding protein and the correlation of RNA-Protein co-expression. For many RNA-Protein pairs the co-detection was as low as 10%, while individual RNA and Protein only detection was higher. The expression of RNA and Protein is often inferred from one another. Therefore we looked into the correlation of RNA-Protein pairs expression. This analysis revealed strong differences depending on if the correlation was computed on the whole sample (heterogeneous population) or single cell levels. Overall RNA-Protein pairs correlation showed greater positive significance on the single-cell level, but higher correlation on a population level. The differences in codetection and correlation of the transcriptome and proteome highlight the importance of multimodal profiling at the single-cell level.
Next, as a proof of concept study, we attempted a biomarker discovery by focusing on melanocytic cells from healthy skin, primary melanoma, and metastatic melanoma. Differential protein expression of surface markers expressed on melanoma cells from metastatic lymph nodes identified unusual expression of CD56 surface marker only on the protein level. Finally, spatial profiling using multiplex immunohistochemistry confirmed the expression of CD56 and revealed its peculiar spatial organization.
In this study, we show that setting manual thresholds is a straightforward way to remove antibody background noise that could otherwise result in false negative interpretations. More computational approaches were introduced recently to remove the false positive noise coming from ADT such as quantification of empty droplets that contain the unbound ABs and normalization using isotype controls2. Using the PBMCs model we demonstrate that the effects of enzymatic dissociation are not limited to changes in gene expression and epitope preservation, but could result in digestion-associated epitope gains in vitro. RNA-protein codetection analysis demonstrated that neither transcriptome nor proteome alone is reliable to conclude the presence of the marker supporting the complementarity of the technology. We also show that optimized CITE-seq is a powerful technology for biomarker discovery but requires spatial validation. Recently, several studies translated the CITE-seq technology into a spatial profiling space3 which adds the additional important biological resolution of the spatial organization of complex tissue and quantitative readouts of molecular landscapes.
- Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
- Mulè, M. P., Martins, A. J. & Tsang, J. S. Normalizing and denoising protein expression data from droplet-based single cell profiling. Nat. Commun. 13, 2099 (2022).
- Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. (2023) doi:10.1038/s41587-023-01676-0.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
Cell death and inflammatory signalling
Publishing Model: Hybrid
Deadline: Oct 28, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in