The origins of the Marcus inverted excited-state dynamics in cobalt(III)
Published in Chemistry

Photoredox catalysis using earth-abundant first-row metal complexes is a vibrant and growing area of research. Despite this, extensive photophysical investigations over the last two decades have predominantly focused on just a few elements, such as iron(II), chromium(III), and copper(I). A significant challenge in developing effective photocatalysts from first-row transition metals is the presence of lower-lying ligand-field excited states. These states tend to deactivate the photocatalytically relevant charge-transfer excited states, with a notable exception of the charge-transfer excited states associated with d10-configured Cu(I).
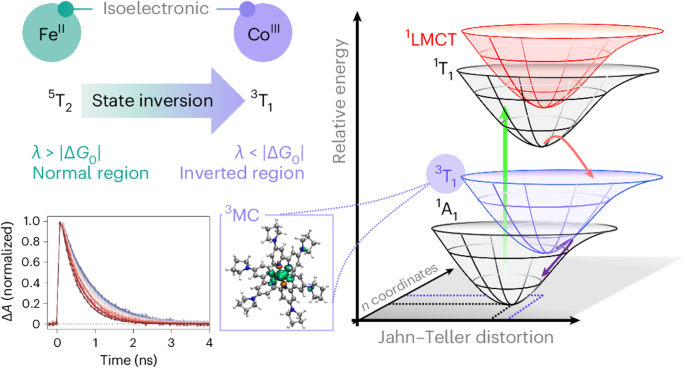
Unlike charge-transfer excited states, ligand-field excited states are not characterized by charge separation but rather a rearrangement of electrons among orbitals localized on the metal center. Initially, this led to the belief that their ability to engage in bimolecular reactivity was limited due to this restricted spatial distribution coupled with the absence of an easily defined chemical potential. However, recent discoveries have shown that these states can indeed exhibit photocatalytic activity. For example, complexes of Cr(III), which have long been known to engage in photo-induced electron transfer, have been utilized in some specific photocatalytic applications aided in part by their relatively long excited-state lifetimes. However, their lowest energy excited state is an intra-configurational ligand-field state which results in minimal geometric distortion from the ground state, leading to a scenario where the energy stored in the excited state cannot be tuned to any significant extent by manipulating the ligand-field strength. In contrast, the excited ligand-field manifold of Fe(II) polypyridyl complexes is very sensitive to the local coordination environment. These states are accessed via rapid (i.e., sub-picosecond) deactivation of metal-to-ligand charge-transfer excited states that dominate their visible absorption spectra, resulting in the formation of a metal-centered inter-configurational ligand-field state. For the vast majority of such compounds, this state derives from a (t2)4(e*)2 configuration, whereas the ground state has a (t2)6(e*)0 configuration. The presence of two electrons in σ* orbitals induces significant geometric distortion, leading to a situation where an increase in the excited-state energy leads to a decrease in its lifetime, This is a major disadvantage for solution-phase photoredox catalysis, where the excited-state lifetime must be sufficiently long relative to the time scale for diffusion - typically on the order of nanoseconds – in order for it to engage in a bimolecular reaction.
The intensity of the MLCT absorption band, while effective for light capture, has the disadvantage of obscuring the much weaker ligand-field absorption which are needed in order to obtain quantitative information about ligand-field parameters. Recently developed Resonant Inelastic X-ray Scattering (RIXS) techniques can address this, but they require access to specialized central beamline facilities and are therefore not broadly accessible. To circumvent this, Dr. Jim McCusker, corresponding author of this paper proposed using isoelectronic cobalt(III) complexes to estimate ligand-field parameters, which could then be translated to other d6-metal based chromophores. This hypothesis proved fruitful as cobalt(III) complexes possess charge transfer bands that are significantly higher in energy, thereby revealing the weaker d-d absorption bands in the visible region of the spectrum. Dr. Jonathan Yarranton from the McCusker research group synthesized a series of cobalt(III) complexes and characterized their d-d absorption properties, enabling the quantification of ligand-field parameters (i.e., 10Dq as well as the Racah B andC).
With the availability of such detailed quantitative information on ligand filed-state energetics, Dr. McCusker sought to explore the excited-state dynamics of these cobalt(III) complexes. I joined McCusker's group in Fall 2020 and eagerly dived into this project, as comprehensive photophysical studies on cobalt(III) polypyridyl complexes were lacking (in stark contrast to the isoelectronic iron(II) complexes which have been extensively studied for over 25 years).
Using ultrafast transient absorption spectroscopy, I measured the ground-state recovery lifetime of the [Co(bpy)3]3+ complex in acetonitrile. Given the stronger ligand-field strength of cobalt(III) (~3,000 cm-1), we expected a decreased in excited-state lifetime compared to the iron(II) analog due to the larger zero-point energy difference. Surprisingly, whereas the [Fe(bpy)3]2+ complex has a ground-state recovery lifetime of approximately 1 ns, I measured a lifetime of about 5 ns for the cobalt(III) analog. This unexpected result prompted further investigation into the excited-state dynamics of these cobalt(III) complexes. Subsequent measurements on a series of cobalt(III) complexes with increasing ligand-field strength revealed that the lifetime increased with increasing driving force, suggesting a contrasting behavior compared to iron(II) photophysics. These results can be explained by invoking the counterintuitive Marcus inverted region, where an increase in driving force decreases the rate constant (and therefore an increase in the excited-state lifetime). This unique photophysical behavior implies that more energy can be stored in the excited states, enabling higher energy chemical processes while also increasing the lifetime—a win-win situation.
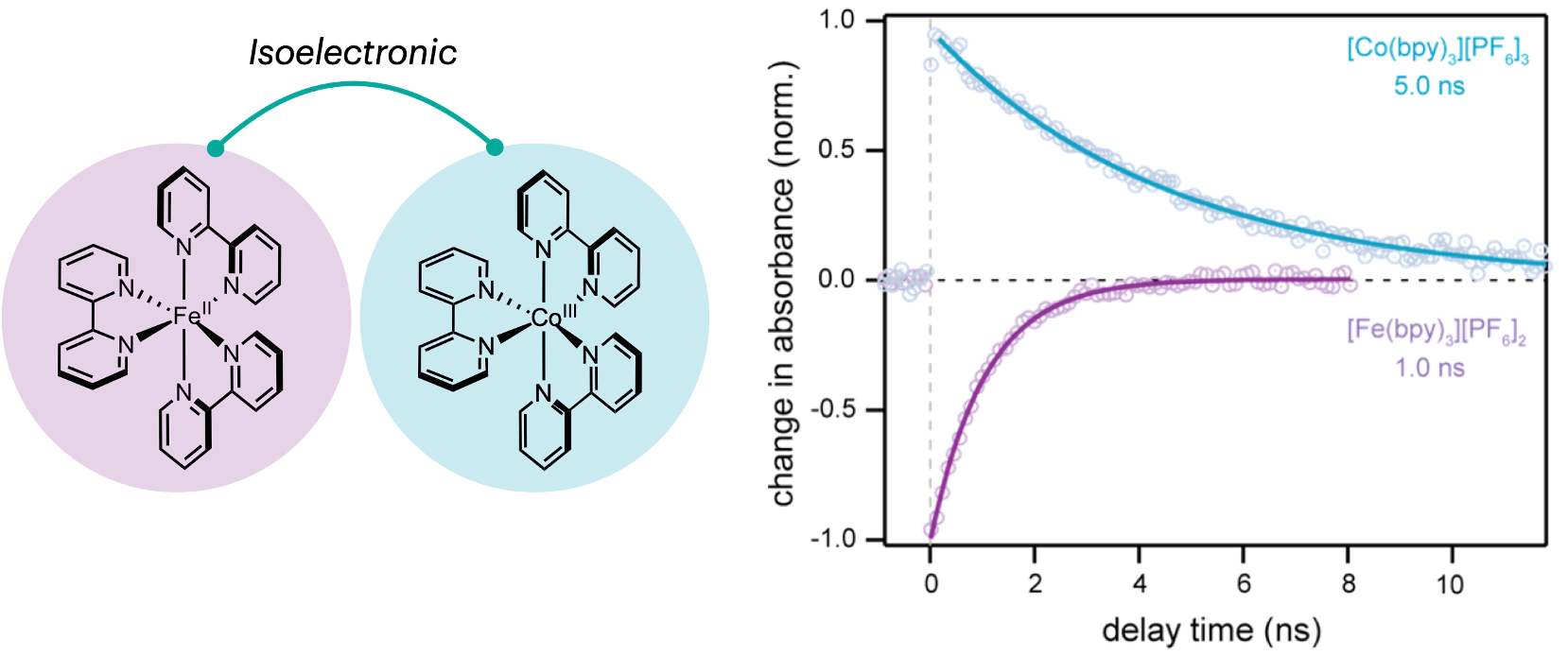
Fig. 1. (left) ChemDraw structures of isoelectronic [Fe(bpy)3]2+ and [Co(bpy)3]3+ complexes. (right) Ultrafast transient absorption decay kinetics showing longer excited state lifetime for the later when measured under identical conditions.
To delve deeper into the origin of this inverted region behavior, in this paper we conducted variable-temperature ultrafast transient absorption experiments on a series of cobalt(III) complexes. The data were analyzed using various non-radiative decay theories, including semi-classical Marcus theory, to quantify reorganization energy and the electronic coupling matrix element that defines the ground-state recovery process. Our findings strongly suggest that the lowest energy excited state for these cobalt(III) complexes is 3T1, characterized by a (t2)5(e*)1 configuration, unlike the 5T2 state of iron(II) complexes, described above. This difference results in a smaller geometric distortion in the excited state of cobalt(III) relative to an Fe(II) analog, leading to a decrease in the total reorganization energy. We concluded that this reduction in reorganization energy, coupled with the increased driving force, pushes the dynamics into the Marcus inverted region. In fact we have recently leveraged this principle for developing a cobalt(III) based photocatalyst to access oxidatively resistive carbon-nitrogen bond forming reactions (see this). This fundamental study provides critical insights that can aid in the development of new chemical platforms utilizing this principle and knowledge on the excited state geometry, opening exciting new avenues for the photoredox community.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in