Peptide synthesis at the origins of life: energy-rich aminonitriles by-pass deactivated amino acids completely
Published in Chemistry

Find our paper Peptide ligation by chemoselective aminonitrile coupling in water by Pierre Canavelli, Saidul Islam & Matthew Powner at Nature (https://doi.org/10.1038/s41586-019-1371-4).
Peptides are an essential element of all life on Earth. They are so highly enmeshed in physiology that it is difficult to imagine life without them. Peptide biosynthesis is now orchestrated by a complex host of genetically encoded enzymes, but it is inconceivable that these sophisticated and coordinated macromolecules suddenly emerged at the origin of life. A simpler scenario must lie at the root of peptide biosynthesis, but can the innate reactivity of prebiotic molecules initially replace enzymes? Can chemical reactivity control selective peptide ligation in water?
Peptides are widely assumed to be products of amino acid polymerization reactions. Whilst conceptually simple, in practice there are good reasons why these reactions are ineffective in water. For example, amino acids (pKaH = 9.4) are zwitterionic at physiological pH, which quenches their nucleophilicity by protonation, and electrophilic condensation agents that activate amino acid monomers can irrevocably block peptide synthesis as well as derivatise amino acid side chain residues.
In this week’s Nature our group has reported a method to overcome several long-standing problems of peptide synthesis in water, which avoids uncontrolled polymerization reactions and by-passes amino acids altogether [1].
We took our initial cues from biology. Whereas chemical peptide syntheses generally proceed from the C-to-N terminus with ligation occurring at the activated monomer, both ribosomal (RNA-mediated) and non-ribosomal (thioester-mediated) peptide biosynthesis proceeds from the N-to-C terminus with ligation occurring at the activated C-terminus of the peptide chain (Fig. 1).

Figure 1 | Peptide synthesis. Chemical peptide synthesis generally proceeds from the C-to-N terminus and requires a two-stage protecting-group strategy with orthogonal protecting groups for the activated monomers alpha-amine (orange) and the amino acid residues (red). Conversely, biological peptide synthesis proceeds from the N-to-C terminus without protecting groups. PG = protecting group, R = amino acid side chain, X = leaving group.
As life has twice alighted on the same solution, it suggested to us that N-to-C-ligation may be privileged, and we suspected it would also be key to achieving prebiotic peptide synthesis in water. Furthermore, prokaryotic and mitochondrial protein syntheses are both initiated by N-formylation, and over 80% of eukaryote proteins are N-acetylated. Peptide N-acylation appears essential to prevent DKP-induced peptide degradation, so it seemed to us that peptide synthesis should proceed from the N-to-C terminus and be initiated by N-acylation.
But what substrates should be ligated and how should the peptide C-terminus be activated? We suspected that aminonitriles were the missing piece of this puzzle.
Aminonitriles have been widely explored as precursors of amino acids [2][3], but their potential in peptide ligation has been largely overlooked. alpha-Aminonitriles have a remarkably low basicity (pKaH = 5.3) and they are only protonated in acidic solution (Fig. 2), which makes them ideally suited to ligation at physiological pH.
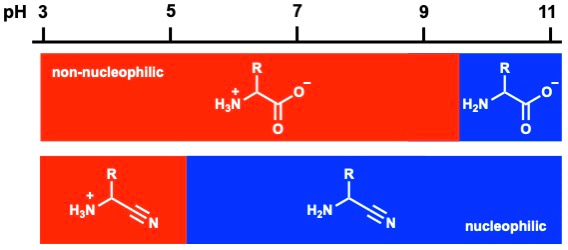
Figure 2 | Nucleophilicity in water. Amino acids are zwitterionic and their nucleophilicity is quenched by protonation at near neutral pH (red), whereas alpha-aminonitriles are only protonated in acidic solution. alpha-Aminonitriles are nucleophilic over abroad pH range (blue), making them ideally suited to peptide ligation at neutral pH.
Additionally, aminonitriles have built-in energy (in their triple bond) that can be harnessed during peptide synthesis, unlike amino acids, which are their deactivated hydrolysis products. Drawing on our previous experience using thioacid activation to direct chemoselective 3'-5'-RNA ligation [4] and nitriles in prebiotic nucleotide synthesis [5][6], as well as recognising the potential commonality between peptide and nucleic acid chemistry, we began to investigate the irresistible opportunity that thioacid chemistry could also ligate alpha-aminonitriles to yield peptides in water.
Upon considering thioacids for peptide synthesis, we were immediately struck by the orthogonal reactivity of phosphates and sulfides, and the opportunities that sulfides opened for selective peptide ligation in a chemical network that needs to simultaneously build peptides and nucleic acids. Lipmann [7] and de Duve [8] had both previously proposed thioester-dependent peptide synthesis preceded RNA-dependent protein synthesis during the evolution of life. This “thioester world hypothesis” is a popular theory in origins of life research, but there had been very little progress in finding a robust prebiotic mechanism to synthesise the thioesters that are required to underpin this whole concept. We suspected this may have been due to a misplaced focus on amino acids and thioesters, rather than on aminonitriles and thioacids.
We found that thioacetate not only quantitatively acetylated alpha-aminonitriles in water, but that acylation electronically activated the nitrile group of the newly formed alpha-amidonitrile just enough to favour nucleophilic attack by H2S, but not by water. The resulting alpha-amidothioamide was then readily hydrolysed to afford a C-terminal amidothioacid which can be ligated with another alpha-aminonitrile, and so the process can be cycled to yield an iterative peptide ligation in water (Fig. 3) [1].
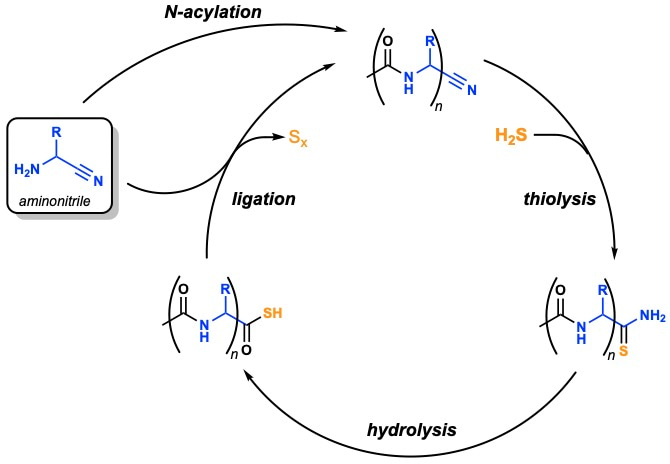
Figure 3 | Peptide ligation cycle. Sulfide-mediated peptide synthesis by N-to-C terminal ligation of aminonitriles in water.
All 20 proteinogenic side chain residues were tolerated (and gave high yields) in amidothioacid ligation, including histidine, aspartate, cysteine, serine, threonine, and tyrosine, which are essential to enzyme catalysis but are notoriously difficult to couple by previously reported (prebiotic) peptide bond forming reactions. The low basicity of alpha-aminonitriles even permits selective ligation of lysine residues to give natural alpha-peptide bonds. This lysine selectivity derives from the ~5 orders of magnitude difference in basicity of the two amine moieties of lysine nitrile. And finally, because our strategy operates via C-terminal peptide activation, rather than monomer activation, peptide fragment ligation is just as effective as monomer ligation by our strategy, which provides a mechanism to rapidly build larger peptides [1].
Our results provide yet another clue that implicates cyanosulfidic chemistry at the origin of life [1][2][4][5][6][9][10], and we are currently developing other variants of our aminonitrile ligation. We are also exploring the emergence of function in these systems to further validate the prebiotic credentials of our chemistry.
Nevertheless, our results already have broad implications for understanding the origins of life. The highly predisposed nature of these reactions, under simple mild aqueous conditions, suggests that the chemistry of cyanide and sulfide foreshadowed enzyme-controlled peptide biosynthesis pathways.
This research by has been published in Nature - please click here to read more.
Behind the paper post, written by Saidul Islam & Matthew Powner, Department of Chemistry, UCL, UK.
References:
[1] Canavelli, P., Islam, S. & Powner, M. W. Nature (2019).
[2] Islam, S., Bucar, D.-K. & Powner, M. W. Nat. Chem. 9, 584–589 (2017).
[3] Islam, S. & Powner M. W. Chem 2, 470–501 (2017).
[4] Bowler, F. R. et al. Nat. Chem. 5, 383-389 (2013).
[5] Powner, M. W., Gerland, B. & Sutherland, J. D. Nature 459, 239–242 (2009).
[6] Stairs, S. et al. Nat. Commun. 8, 15270 (2017).
[7] Lipmann, F. Science 173, 875–884 (1971).
[8] de Duve, C. Blueprint For a Cell: The Nature and Origin of Life (Neil Patterson Publishers, Burlington, NC) (1991).
[9] Patel, B. H. et al. Nat. Chem. 7, 301–307 (2015).
[10] Roberts, S. et al. Nat. Commun. 9, 4073 (2018).
Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in