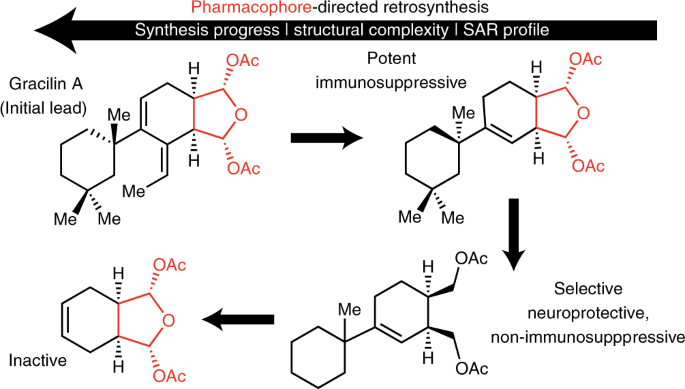
Pharmacophore-Directed Retrosynthesis: Intertwining Natural Product Total Synthesis and Structure-Activity Relationship Studies
Published in Chemistry
Synthetic chemists continue to be inspired by small molecules isolated from various natural sources, including plants, bacteria, and marine sponge to name a few. The bioactivity of these compounds has always been of interest to chemists, but when it comes to devising a retrosynthesis (working backwards from natural product to simple starting materials), the focus is typically on identifying strategic bond disconnections that lead to the most efficient total synthesis (fewest steps). For many decades, this has driven the field of synthetic chemistry forward by encouraging novel bond disconnections and streamlining synthetic routes. Synthetic chemists continue to be intrigued by the biological activity of the molecules they are targeting for total synthesis. Efficient synthetic routes enable biological investigations that delve further into trying to uncover the rich biology that can be learned through the synthesis and detailed structure-activity relationships (SAR) of natural products and derivatives including simplified versions with comparable or possibly unique biological activity.
In the 1980’s and 90’s, the groups of Danishefsky, Schreiber, and Wender, for example, building on historical examples by Corey and Woodward among others, began to integrate their interests in natural product total synthesis with the biological activity of these complex small molecule targets. In our first NIH grant application, we targeted the synthesis of the marine natural product, pateamine A, and proposed biological studies, including cellular receptor isolation following completion of the total synthesis (I think this was a common theme in many grant applications to NIH in the early 1990’s). We completed the total synthesis of pateamine A (PatA) in 1998 (JACS, 1998), and, building on a hypothesis regarding binding and scaffolding domains of this natural product, we hypothesized that we could remove two stereocenters (out of four total) and thus greatly streamline the synthesis. Indeed, the synthesis of des-methyl, des-amino pateamine A (DMDA PatA) went from a 24-step sequence for the natural product to a 14-step sequence (JACS, 2004). Importantly, this simplified derivative was nearly equipotent to the natural product, retaining its activity as a nanomolar protein translation initiation inhibitor. DMDA PatA has served as a useful chemical probe for biologists studying translation initiation (>60 samples sent to labs worldwide). There are, in fact, many recent examples of truncated natural products leading to cellular probes but also interesting lead compounds for drug discovery. One of the most dramatic examples may be the story of halichondrin, which, after truncating nearly half of this highly complex molecule, led to the FDA-approved drug, Halaven® (eribulin mesylate) in 2010 (NatProdRep, 2017). Our own work with pateamine A led to the retrospective question of why we had not targeted the synthesis of DMDAPatA en route to the natural product itself? This line of thinking ultimately led to the concept disclosed in this paper; namely, the development of a retrosynthetic strategy premised on a hypothesized pharmacophore. Application of PDR can lead to several simplified versions of the natural product bearing the hypothesized pharmacophore en route to the natural product and importantly it enables biological testing to commence at a much earlier stage in a total synthesis effort.
In this paper, we describe the first application of this strategy to the marine sponge-derived from the natural product, gracilin A, which had recently been re-isolated (including congeners) by Prof. Marcel Jaspars (Univ. of Aberdeen, Scotland) and supplied to our collaborator, Prof. Luis Botana (Univ. de Compastela, Spain) who discovered that this natural product exhibited immunosuppressive and neuroprotective activity. Our recent synthesis (ChemEurJ, 2015) and proteomic studies (ChemComm, 2017) of a related natural product, spongiolactone, led Prof. Jaspars to contact us and put us in touch with Prof. Botana to determine if we wanted to delve into a total synthesis of gracilin A. This came at a perfect time as we had just started formulating the idea of PDR and we were seeking a proof-of-principle study for this synthetic strategy. Currently, we are continuing to tease out the factors that lead to selectivity between cyclophilin A and D and thus immunosuppressive versus neuroprotective effects through molecular modeling and further derivative synthesis.
It should be noted that in cases where the natural product is readily available either through isolation as a major congener or through fermentation, the application of PDR may even preclude the need to complete a total synthesis. Now, reading this may cause some concern in the minds of some of my hard-core total synthesis colleagues, since this may be seen as a convenient exit point for graduate students out of a total synthesis effort. I understand this sentiment wholeheartedly, but I am pleased to find that most of my current students are still driven to complete a total synthesis of their natural product no matter what biological discoveries might be made along the way!
Finally, pharmaceutical companies looking once again to natural products as drug leads may find this strategy very appealing. PDR could mitigate some of the concerns with the complexity of natural products which contributed to the decline of natural products as starting points for drug discovery in the first place!
You can read more about Pharmacophore-Directed Retrosynthesis at Nature Chemistry.
This blog was co-authored by Christian M. Chaheine and Daniel Romo.
Poster Image was designed and created by Mikail E. Abbasov.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in