Photophysics of single-molecule magnets
Published in Chemistry

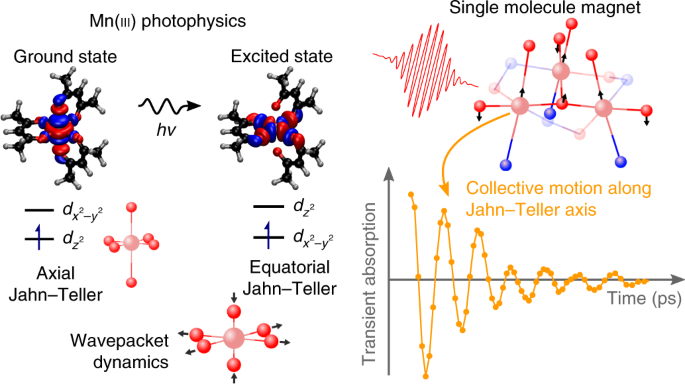
I first came across the field of ultrafast magnetism in 2013 when I was looking for ideas to start my independent research career. By then, the field had grown rapidly since the discovery in 1996 by Jean-Yves Bigot and Eric Beaurepaire and co-workers that femtosecond (fs) laser pulses could demagnetize ferromagnetic nickel films in less than 100 fs. These finding were completely unexpected and there are still many unanswered questions today. The initial experiments were carried out using a polarization-sensitive pump-probe technique. The ultrafast magnetisation dynamics could be detected by recording the change with time in polarization direction of the probe beam as a result of the magneto-optical Kerr effect (MOKE) (or Faraday effect, depending on the geometry of the experiment). Since then,there have been numerous studies and it is now even possible to completely reverse the magnetisation direction using only fs laser pulses. I was amazed by these results and fell in love with the possibility to use fs laser pulses to both initiate and “see” magnetisation precession in materials. What became clear was that there had been no studies on molecular magnets using these techniques. I was therefore wondering if it would be possible to apply some of these time-resolved magneto-optical techniques to study molecular magnets.
We thought that it would initially be best to study so-called molecule-based magnets for the magneto-optical measurements. These are coordination polymers made from molecular building blocks that form a 3D network. I chose to study a Prussian blue analogue because of the possibility to make transparent thin films using electrochemistry, which seemed suitable for the measurements. Since I was just starting my lab, I contacted Jean-Yves Bigot at IPCMS in Strasbourg to see if we could do the MO measurements together in his lab. Jean-Yves had already thought about molecular magnets so was keen to start a collaboration. After a few weeks travelling back and forth, we managed to observe the first ultrafast magneto-optical signal from a molecule-based magnet. Jiwan Kim, who was a postdoc in Jean-Yves’ group, and I did the experiments together during some very intense couple of weeks. This was a very exciting time and I’m very proud of what we accomplished.
Of course, the goal is to reach the molecular limit and study the magnetization dynamics in single-molecule magnets (SMMs) instead of coordination polymers. These are polynuclear metal-coordination clusters in which unpaired electrons on ions in the complex couple to produce a high-spin ground state with an energy barrier between the “up” and “down” spin alignment along some internuclear axis. I therefore started a collaboration with my colleague Euan Brechin, who is an expert in making SMMs. It was natural to start with one of his molecules, [Mn(III)6O2(Et-sao)6(O2CPh(Me)2)2(EtOH)6], or “Mn6”, which had a record anisotropy barrier for a Mn-based SMM. In Mn6, six high-spin Mn(III) ions are arranged in two triangles. The electron configuration t2g3eg1 leads to a Jahn-Teller (JT) distortion, which in turn leads to zero-field splitting and SMM behaviour. From a photophysics point of view, this large molecule seemed quite unwise to study and not much work has been done on such large systems. One reason for this is that with open-shell systems (unpaired electrons), it is difficult to do reliable quantum chemical calculations of excited states. Nevertheless, we decided to try and see what we could find. Our approach was initially to compare a complex containing three Mn(III) ions in one triangle [Mn(III)3O(Et-sao)3(β-pic)3(ClO4)], or “Mn3“, with the two triangles in Mn6.
At the same time, my PhD student Florian Liedy started in the group and he wanted to work on SMMs. Before doing any magnetic measurement, we thought the first measurement should be “standard” transient absorption (TA) to explore the photophysics. In fact, one of Florian’s first tasks was to build a TA spectrometer and he did a great job. We did not expect to see anything interesting based on the featureless absorption spectra of both Mn3 and Mn6. However, rather unexpectedly, we observed some structure in the TA spectra. There were even some subtle differences between Mn3 and Mn6, which lead us to believe that the JT distortion could be involved in the dynamics. We tried many different measurements and as Florian went on to improve the apparatus, we could achieve a very good signal-to-noise ratio and one day I came in to the lab and we could see oscillations in the decay traces. This was a very exciting finding and there was a real buzz in the lab. By collaborating with Julien Eng and Tom Penfold (Newcastle University), we could identify the vibrational motion giving rise to the wavepacket in Mn3 and indeed identify a collective motion along the JT axis. We wrote up the paper and submitted it to Nature Chemistry.

Figure 1: Part of the transient absorption spectrometer that Florian Liedy built.
We eventually got back the reviewer comments and one of the suggestions was to go to an even smaller system, namely a monomer. This was a great idea but there was no obvious candidate, in particular with SMM properties. Ever since I started working in the field of inorganic photophysics, I have been inspired by Jim McCusker’s work (in particular the work on Cr(acac)3). It therefore seemed obvious to try Mn(acac)3. Funnily enough, we had gone from Mn6 to Mn3 to Mn(acac)3, although with hindsight we should have gone the other way around. It was really interesting to see that we could observe the same transient absorption spectra and even some oscillations in the Mn(acac)3 data. The oscillations were different and much less pronounced than in Mn3. Again, it was a very nice surprise that we could see a clearer signature of the wavepacket in the larger molecule. However, the smaller size of the monomer meant that Julien and Tom could carry out high-level quantum chemical calculations and identify the electronic states giving rise to the wavepacket motion. So I guess one of the main lessons I learned was don’t be afraid to try crazy ideas but make sure you find a good, simple model system to be able to make sense of the more complex system.
From my interest in ultrafast magnetism in condensed matter, it was clear that anisotropy was important. This is why I find the observation of a coherent wavepacket along the JT axis in an SMM so interesting. In next-generation hard drives, lasers are used in the heat-assisted magnetic recording (HAMR) technology currently being developed by Seagate, to transiently reduce the magnetic anisotropy in a thermal mechanism. Our results could enable the use of quantum coherence instead of a thermal mechanism and will therefore allow for terahertz operational rates instead of gigahertz with the additional benefit of the zero-dimensional nature SMMs. Whether SMMs will ever be used in data storage is still an open question, but with recent developments of SMMs with blocking temperatures close to or above liquid nitrogen, this is becoming a stronger possibility.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in