Predicting sequence evolution through machine learning
Published in Bioengineering & Biotechnology


Genome sequencing surveys have unraveled previously unfathomable degrees of sequence diversity. Beyond point mutations (or SNPs), massive amounts of sequence rearrangements were identified thanks to innovations in sequencing technology and assembly algorithms. Long-read genome assemblies make it now possible to assemble most prokaryote and smaller eukaryote genomes with ease. We are just at the cusp of getting fully resolved human chromosomes.

Disease symptoms caused by the pathogen on wheat leaves. Public domain picture by Maccheek (Wikipedia)
With the progress in getting complete genomes, Thomas Badet, a junior group leader at the University of Neuchâtel and I started wondering how we could capture the full extent of chromosomal rearrangements segregating within a species. In our paper, just published in Nature Communications, we mostly focus on a fungal pathogen called Zymoseptoria tritici,which can severely affect wheat production worldwide. We started with defining the pangenome of the species based on representative strains across the world. Comparing genomes, we found an astonishing number of chromosomal rearrangements many of which were affecting genes. As a consequence of the rearrangements, strains carried substantially different sets of genes. We do know now that many of these genes carry out important functions to resist pesticides or cause disease on wheat. Hence, understanding the nature of these gene presence-absence variation is important.
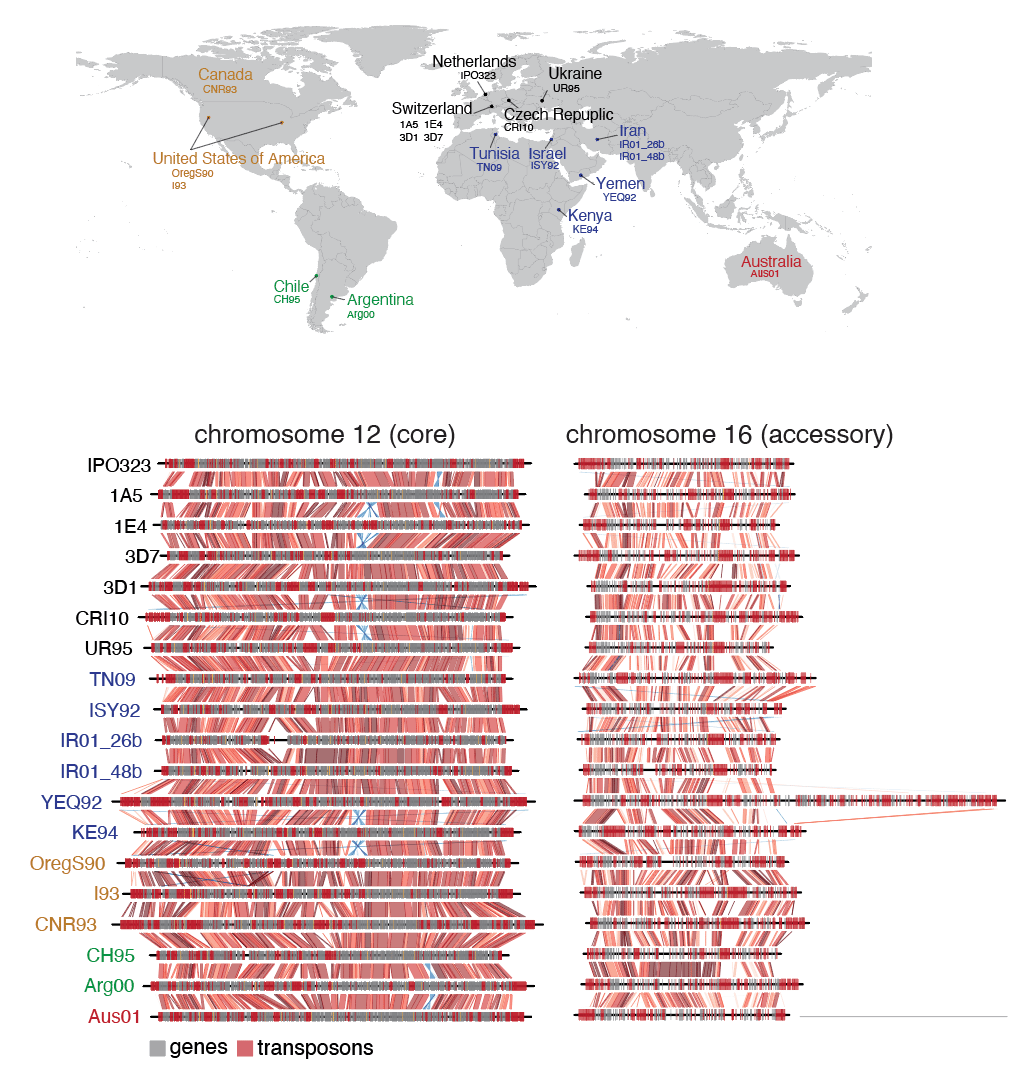
Extensive variation in chromosome structure across the species range.
Given the massive scale of sequence rearrangements, we were wondering whether we could spot patterns where such rearrangements occur along chromosomes. Obviously, repetitive sequences were more likely associated with some form of rearrangement. But what about sequence composition? Epigenetic modifications? Recombination rates? Hence, we started putting together a set of 30 distinct features characterizing individual chromosomal segments. Using machine learning, we train models that were capable of predicting where sequence rearrangements happened. For this, we used a subset of the 19 completely assembled genomes of the species, trained the model and then tested whether we could predict chromosomal rearrangements not included in the training. To our surprise, this worked fairly well.

Machine learning and prediction of chromosomal rearrangements.
To take a step further, we wanted to put our trained model to the test for rearrangements happening in future generations. We established controlled crosses of parents with fully assembled genomes. We have also assembled complete genomes of haploid progeny over multiple generations to track any spontaneous sequence rearrangement. Finally, we applied our previously trained model to predict the appearance of these new sequence rearrangements. Our model performed very well in particular for insertion/deletion variants but was capable of predicting all kinds of rearrangements with at least with some success. To further validate our machine learning approach, we have analyzed a set of eight completely assembled Arabidopsis thaliana genomes. Even for these substantially larger genomes, machine learning showed significant power to predict the location of sequence arrangements in genomes not included in the training.
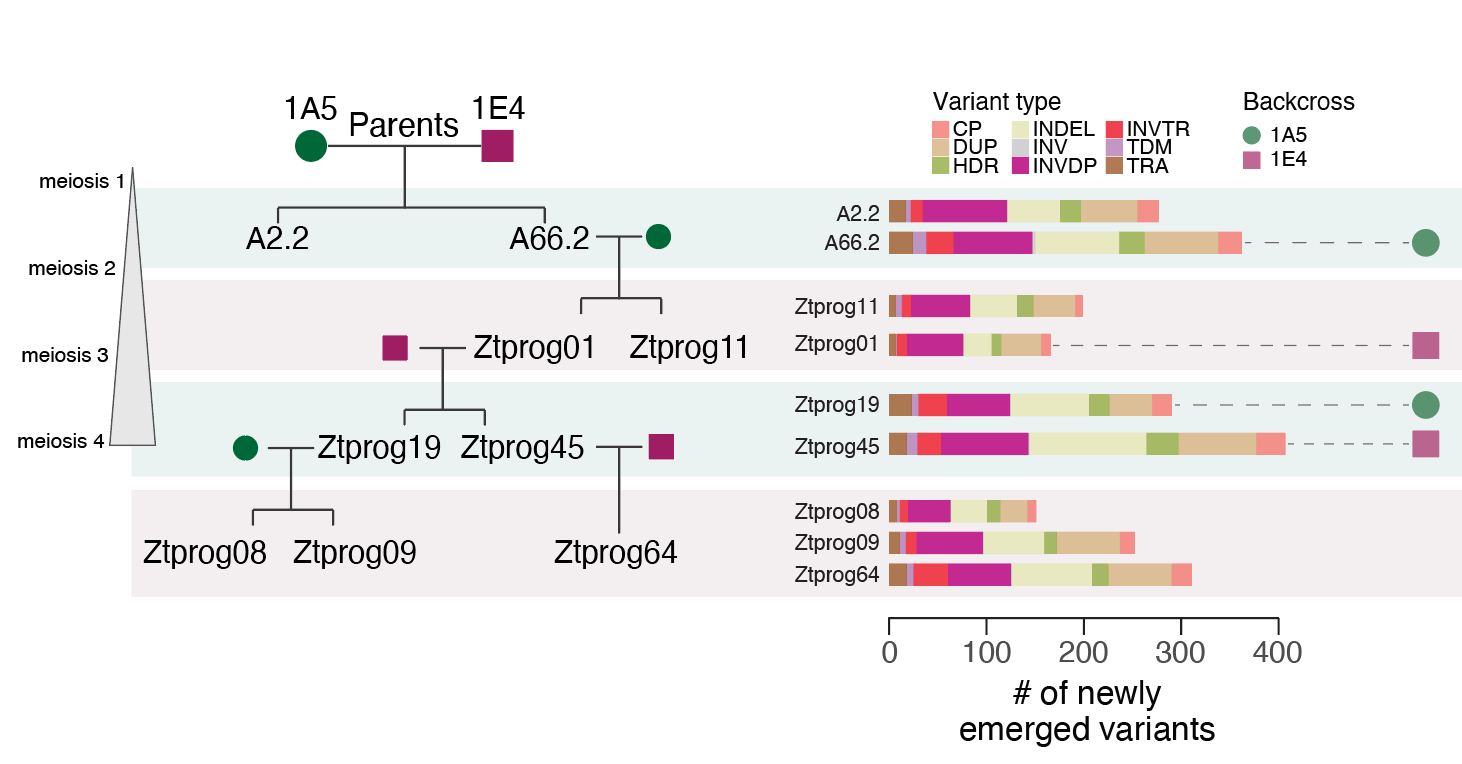
Four-generation haploid pedigree to evaluate the predictive power of the machine learning approach.
Could machine learning also help to predict variation among human genomes? The idea to predict fragile sites prone to rearrangements has received considerable attention in cancer research or heritable diseases. If genomes are of sufficient quality, machine learning could help predict the mutational trajectory of cancer lineages and possibly identify particular weaknesses for treatment. Predicting sequence rearrangements could also help make antimicrobials more durable or genetic modifications in breeding more robust. Much road needs to be travelled still but we believe that machine learning of sequence features will find many future implementations.
Reference
Badet T, Fouché S, Hartmann FE, Zala M, Croll D. 2021. Machine-learning predicts genomic determinants of meiosis-driven structural variation in a eukaryotic pathogen. Nature Communications. DOI: 10.1038/s41467-021-23862-x
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in