Profiling the natural context of antibiotic resistance in microbial communities.
Published in Microbiology
Explore the Research

Linking the resistome and plasmidome to the microbiome - The ISME Journal
The ISME Journal - Linking the resistome and plasmidome to the microbiome
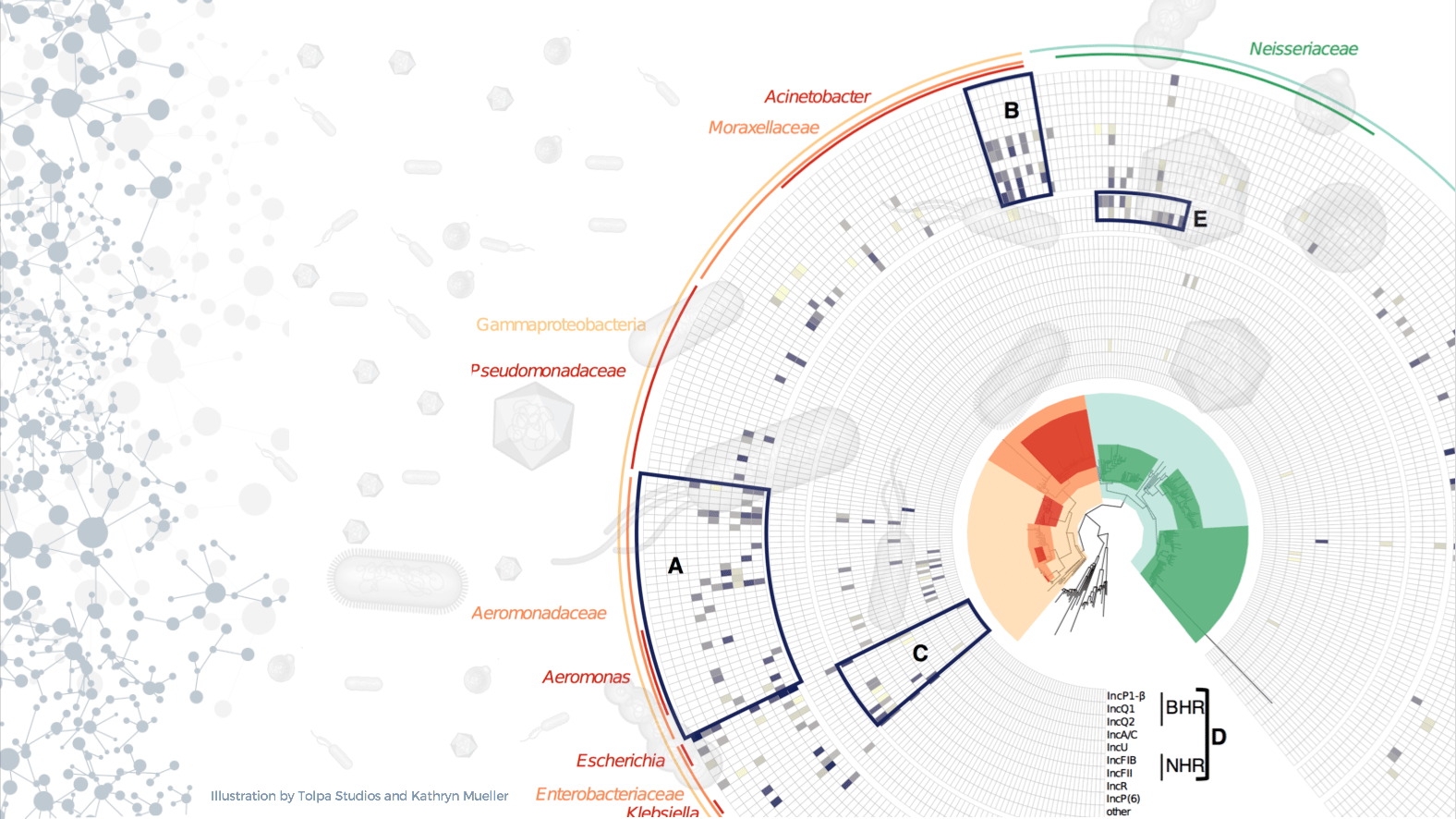
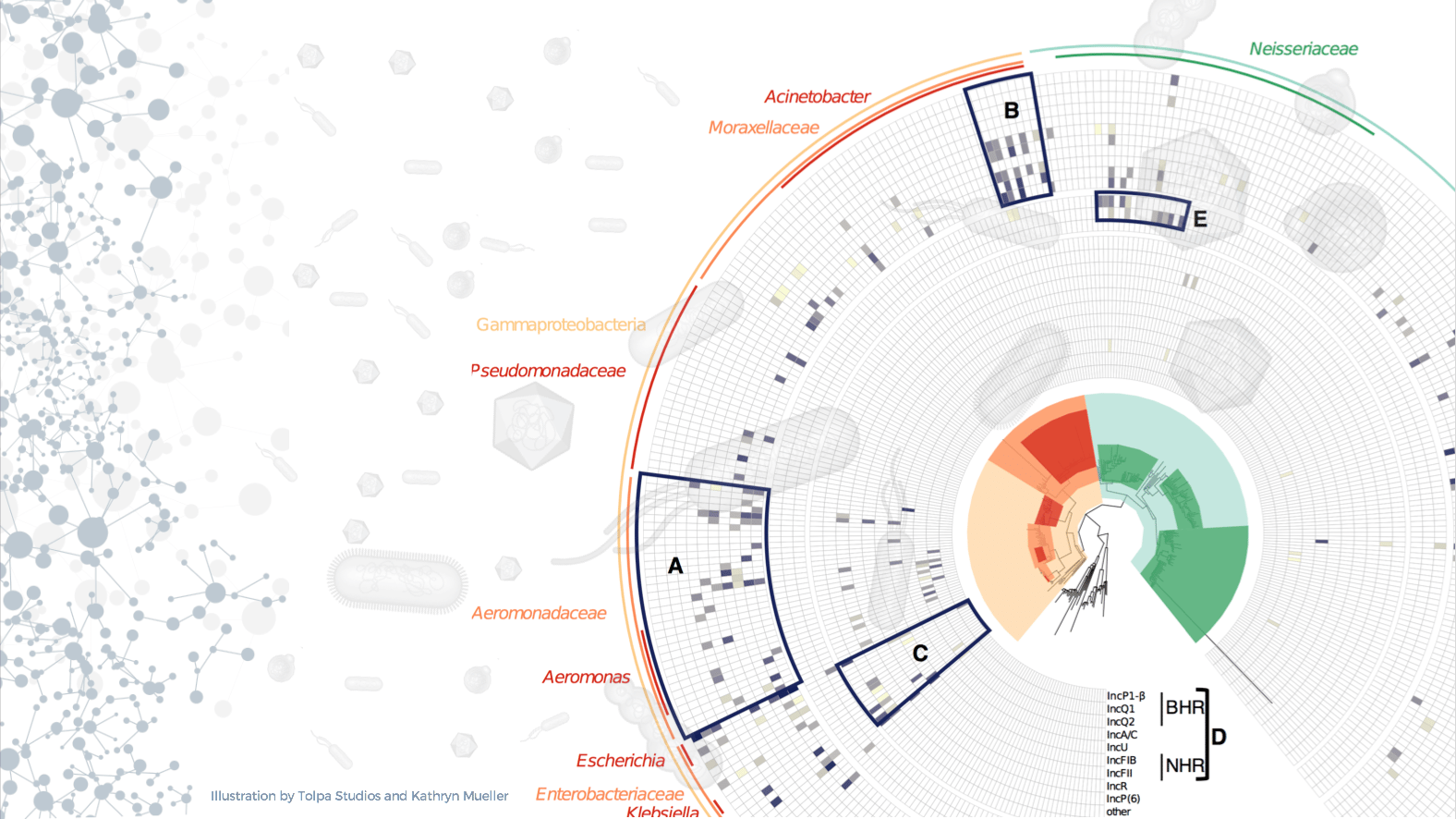
One of the main bottlenecks in the study of complex microbial communities is our inability to track mobile genetic elements without having to isolate individual organisms or cells. Data collected by metagenomic approaches typically provide limited information because of the destructive nature of DNA sequencing. Since mobile genetic elements (plasmids, phage, etc.) are very abundant in microbial settings, this obstacle leaves a big blind spot in our understanding of microbial ecology. Antimicrobial resistance genes are one particular area of interest because they often travel across a population of diverse microbes on plasmids and other mobile genetic elements. If we can profile the natural context of antibiotic resistance determinants, we can learn how resistance emerges and disseminates, and eventually, we may be able to predict which antibiotic resistance genes and plasmids might be found in future infectious disease outbreaks.
This story is the product of a collaboration between the Seattle-based biotech company Phase Genomics and our team at the University of Idaho. Here we present a new approach to determine the natural reservoirs of antibiotic resistance in complex microbial communities. We used the in vivo proximity ligation method Hi-C to detect co-occurrence of antibiotic resistance genes, mobile genetic elements, and chromosomal taxonomic markers by physically linking them before cell lysis. This allowed us to identify the carriers of various antibiotic resistance genes, integrons, and plasmids in a complex wastewater microbiome.
We chose to test a wastewater microbiome sample because they are diverse and complex, and known to be a reservoir of antibiotic resistance. With the large amount of new information gathered in one sample, it was hard to stay focused on our specific question. For example, we did not address the reassembled genomes, 50 of which qualified as basically complete, and more than half of them did not seem similar to any known bacterial species. This tells us that there is so much left to discover under our feet; it feels like spatial exploration at the microscale.
We are just beginning to see the potential of proximity-ligation for microbiome research and the method needs further validation to assess its boundaries and the significance of the results. For that reason, our team and others are working on defining these limitations. This technology will have a significant impact on our ability to determine the sources of antibiotic resistance found in the clinic. It will also be a great tool in any microbiome research effort where one wants to more accurately associate functional genes and mobile genetic elements with their bacterial hosts and improve genome assembly based on whole community DNA sequence data.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in