Programmable late-stage C−H bond functionalization enabled by integration of enzymes with chemocatalysis.
Published in Chemistry

There is an urgent need for more sustainable ways of building molecules to create pharmaceuticals and other products of value to society. Currently multistep processes are often required to make these molecules, utilising expensive and deleterious reagents that create considerable waste, which is damaging to the environment.
Nature has evolved enzymes, biocatalysts, with exquisite selectivity, which enable the multistep assembly (biosynthesis) of molecules to be performed in cells under benign aqueous conditions. Unfortunately, nature does not provide all the biocatalysts required to assemble many of the non-natural products we need today, such as pharmaceuticals.
We reasoned that construction of valuable synthetic targets could be achieved most efficiently and cleanly by combining the selectivity of biocatalysts with non-natural catalysts. For example, synthetic metal catalysts have been developed that can install a wide range of functionality into molecular scaffolds. Whilst powerful, metal catalysts often lack the selectivity afforded by enzymes. If the diverse functionality provided by metal catalysts could be combined with the exquisite selectivity of enzymes, new and more direct synthetic routes might be realised.
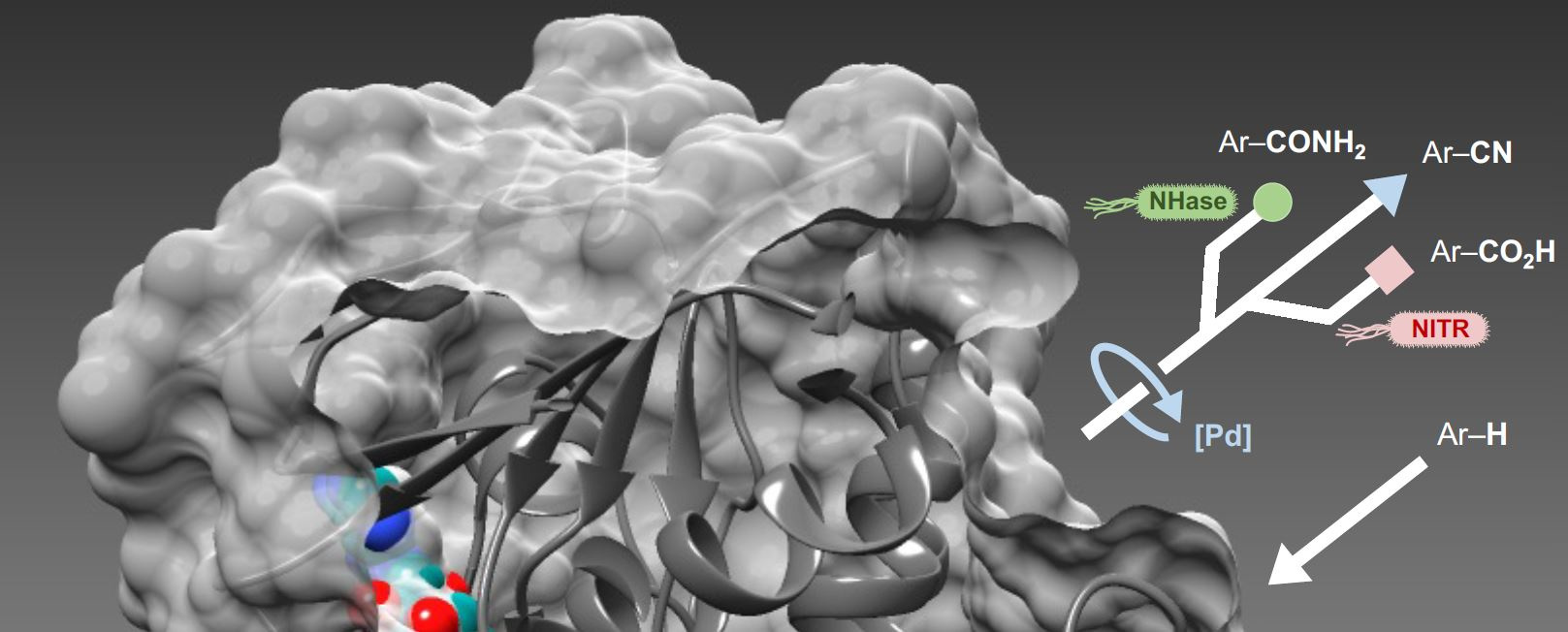
In this study, we aimed to selectively transform ubiquitous carbon-hydrogen (C-H) bonds into new C-C bonds, within aromatic molecules. These are commonly used to manufacture pharmaceuticals, agrochemicals, and other important products. Our strategy involves selectively introducing nitrile groups (CN) to create new C-CN bonds. Nitrile groups are present in many target molecules and can also be transformed into a range of other desirable functional groups.
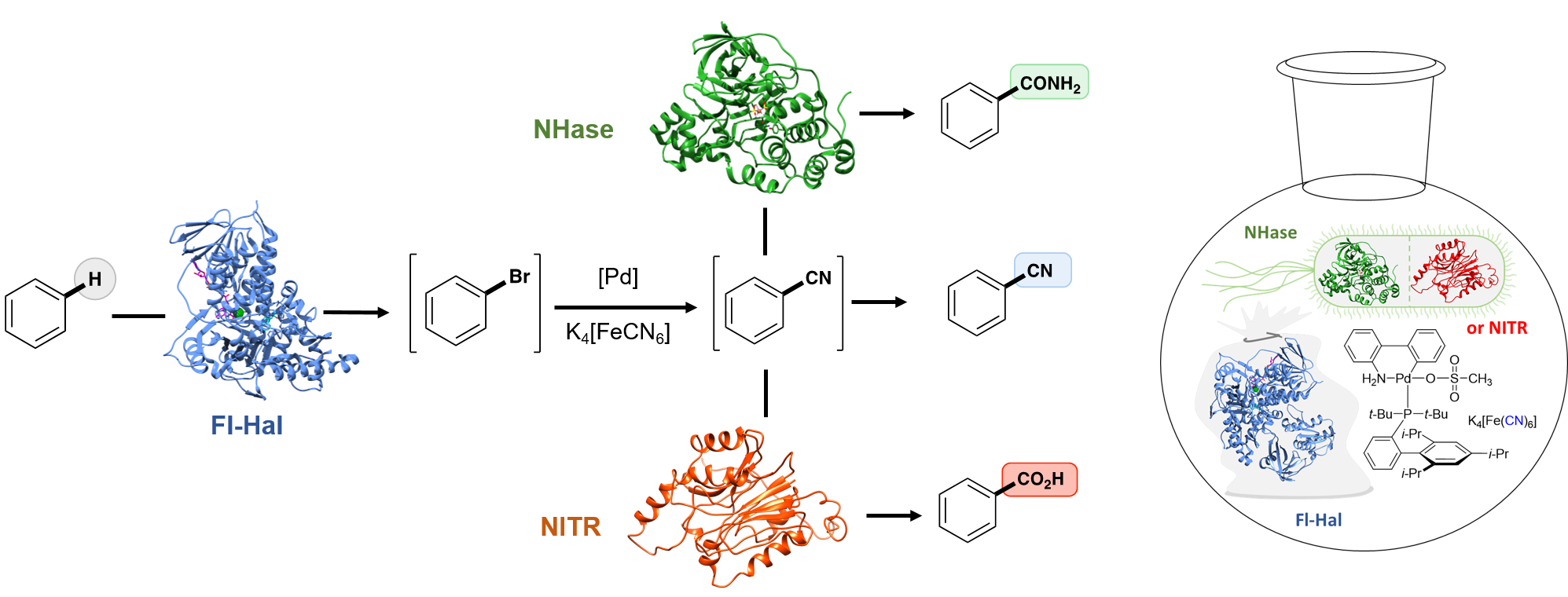
Whilst extremely versatile, the nitrile group is rare in nature and there are no enzymes available that can introduce nitriles into aromatic molecules. In light of this, we chose to use enzymes that transform C-H bonds into new carbon-halogen bonds. Unlike nitrile groups, halogens such as chlorine (Cl) and bromine (Br) are commonly found in natural molecules and nature has evolved a wide range of enzymes (halogenases) that can transform specific C-H bonds into C-Cl or C-Br bonds. Selectivity achieved by these enzymes is often elusive using state-of-the-art chemocatalysis.
With many natural and engineered halogenase enzymes in hand we needed to develop a way of transforming the more reactive C-Br bond into the required nitrile functionality (C-CN). To do this we turned to synthetic catalysis and after extensive screening we found a palladium metal catalyst that could replace the bromine atom with a nitrile group under the aqueous conditions required for the halogenase to function. In this way, we were able to selectively functionalise C-H bonds, first to bromide and then to nitrile in a single ‘one-pot’ reaction process, thus combining two steps into one.
We then sought to transform the nitrile into other useful functionalities. Firstly, we added a nitrile hydratase (NHase) enzyme to the system, which converts nitriles to important amide (CONH2) groups. However, the NHase is deactivated by the presence of the metal catalyst. To overcome this, we added bacterial cells that produce the NHase instead. In this case the cell wall protects the NHase, inside the bacteria, from other components of the reactions, thus allowing us to combine three steps in one-pot. Similarly, cells possessing nitrilase enzymes (NITR) could also be added to yield valuable carboxylic acid (COOH) functionalities.
Currently we are working on extending this approach, combining a range of different enzymes, synthetic catalysts and cells, in one pot processes, to telescope routes to other drugs and other target products.
This research has now been published in Nature Catalysis.
https://www.nature.com/articles/s41929-021-00603-3
Elliott J. Craven, Jonathan Latham, Sarah A. Shepherd, Imtiaz Khan, Alba Diaz-Rodriguez, Michael F. Greaney and Jason Micklefield.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in