Proteogenomic analysis of human pancreatic ductal adenocarcinoma
Published in Cancer
Recent genomic and transcriptomic analyses of human pancreatic ductal adenocarcinoma (PDAC) tissues have identified genomic alterations and genes with cancer-related altered expression1. In addition to the genomic and transcriptomic analyses, proteomic analysis of human PDAC samples and healthy tissues has recently identified proteins with altered expression2,3. However, PDAC-associated proteomic signatures have not been systematically characterized at the global level in large cohorts of patients with PDAC. Integrative analysis of the genome, transcriptome and proteome, known as the proteogenomic analysis, can provide a more comprehensive understanding of the molecular signatures associated with cancer pathogenesis4,5,6,7. Here, we present a proteogenomic analysis of PDAC.
We collected tumor and matched blood samples from 196 patients with PDAC (57, 101 and 38 samples from stages I-III, respectively). Whole exome sequencing (WES) was performed for tumor samples and peripheral blood mononuclear cells (control) isolated from the matched blood samples. The tumor samples were subjected to mRNA sequencing (mRNA-seq). We selected 150 of 196 tumor samples with high tumor cellularity for global proteome and phosphoproteome profiling using liquid chromatography–tandem mass spectrometry analysis.
Key findings
1. Mutation-phosphorylation correlation reveals mutation-signaling interplays in PDAC
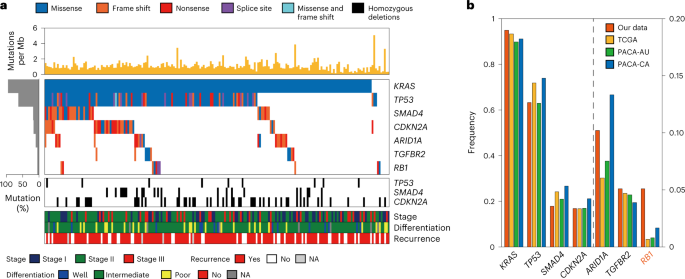
From the WES data, we identified 12,116 somatic mutations affecting protein sequences. Based on these mutations, we identified seven significantly mutated genes (SMGs). Among the seven SMGs, six (KRAS, TP53, CDKN2A, SMAD4, ARID1A and TGFBR2) were shared with the previously identified SMGs8,9,10,11,12, while RB1 was not. We then examined correlations between somatic mutations and protein/phosphorylation levels to infer functional implications of these mutations. Somatic mutations in KRAS, TP53, ARID1A, TGFBR2/SMAD4 and RB1/CDKN2A significantly correlated with increased levels of phosphopeptides for proteins that were associated with RhoA signaling and pathways related to cell proliferation/apoptosis and cell adhesion/cytoskeleton. These data suggest that frequent mutations in PDAC can be associated with the regulation of apoptosis and actin cytoskeleton dynamics.
2. mRNA-protein abundance correlation reveals potential prognostic biomarkers in PDAC
We next examined the correlations between mRNA and protein abundances across samples based on 6,959 protein-coding genes, with both protein and mRNA abundance data available for at least 50% of the samples. Significant positive correlations between mRNA and protein abundances were found for 49.7% of the samples, with an average correlation of 0.42, similar to those previously reported13.
Genes with significant mRNA–protein correlations showed a greater association between their abundance and patient survival. Among the 3,461 genes with significant mRNA–protein correlations, we selected 321 genes with significant negative correlations with survival. These genes tended to be related to pro-tumor processes linked to invasive behaviors. By performing the same survival analysis using mRNA data from three previously reported cohorts8,14,15, we identified 19 genes that showed significant negative correlations with survival in all four cohorts and suggested these genes as potential prognostic biomarkers. For the four of them (PPL, TPI1, PTGES and DCBLD2), we further validated their cell proliferation- and cell migration-related functions based on short hairpin RNA-based knockdown experiments in AsPC1 and PANC1 cell lines. These cell-based assays support the feasibility that they could serve as potential prognostic biomarkers.
3. Integrated analysis of mRNA-protein data identifies six PDAC subtypes
Previous studies1,8,9,16,17,18 defined RNA data-based PDAC subtypes as: (1) ‘squamous’, also called basal-like or quasi-mesenchymal; (2) ‘pure classical progenitor’, also called classical or progenitor; (3) ‘immunogenic progenitor’, also called immune classical; and (4) ‘exocrine-like’, also called aberrantly differentiated endocrine exocrine (ADEX). To investigate whether proteomics data can refine these conventional subtypes, we performed clustering analysis of 196 samples using mRNA-seq data and 150 samples using proteomics data. For the mRNA, protein and phosphorylation data, we identified three, five and five clusters, respectively. Next, we performed an integrated clustering of 150 samples by combining the three clustering results and identified six subtypes (Sub1-6). Sub2–4 showed decreased survival compared with Sub1, Sub5 and Sub6; Sub4 showed the worst median survival of approximately 20 months, which could be predicted by the protein signatures. These data suggest that protein/phosphorylation data further extended mRNA data-based subtypes due to their complementary nature.
4. PDAC subtypes are associated with distinct cellular pathways
We performed functional enrichment analysis for genes and proteins that defined each subtype and identified cellular processes associated with Sub1-6. Genes defining Sub2–4 represented epithelial mesenchymal transition (EMT)-related pathways (e.g., Tgfb/Wnt signaling). Proteins defining Sub2 and Sub3 represented the same EMT-related pathways. However, proteins defining Sub4 more strongly represented cell-cycle-related pathways than those for Sub2–3 (e.g., DNA replication/repair). These results suggest that Sub2–4 share invasive characteristics; however, Sub4 had a higher proliferation capacity than Sub2–3. Genes defining Sub5–6 represented immune-related pathways (e.g., T cell receptor signaling). Correspondingly, proteins for Sub5 represented the same immune-related pathways. However, proteins defining Sub6 strongly represented exocrine-related processes (e.g., amino acid synthesis). These results suggest that Sub5–6 share immunogenic characteristics, although Sub6 was associated with a higher exocrine function than Sub5. Finally, the genes and proteins defining Sub1 represented carbohydrate/lipid metabolism, a previously reported representative feature of classical progenitors9.
We then performed deconvolution analyses to infer cell-type compositions from bulk mRNA-seq data based on previously reported mRNA signatures17,18,19,20,21,22. Sub2-4 comprised a high proportion of basal-like tumor cells, whereas Sub1 showed a high percentage of classical tumor cells, consistent with their high tumor purity and a high proportion of ductal cells. In contrast, Sub5-6 contained a high proportion of immune cells. Sub5 also contained a high percentage of stromal cells, which may be immune-rich. However, Sub6 had high percentages of exocrine cells. Based on these results, we named tumor cell-enriched Sub1-4 as tumor subtypes 1-4 (TS1-4) and immune-rich Sub5-6 as immune subtypes 1-2 (IS1-2). We next classified the samples using the previously reported subtype signatures9,16,18 and observed that TS1 corresponded to the pure classical progenitor subtype and IS1–2 corresponded to the immunogenic progenitor and exocrine-like subtypes, respectively. However, our proteomic signatures further categorized the conventional squamous subtype into activated stroma-enriched invasive squamous TS2, invasive squamous TS3 and invasive and proliferative squamous TS4, providing a unique context for patient stratification.

5. High level of tumor infiltration of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in aggressive TS4 tumors suppresses T cell proliferation
Besides the low expression of B and T cell markers in TS2–4, the levels of neutrophil markers were noted to be higher in TS2–4 than in IS1–2, suggesting potential interactions between myeloid cells and lymphocytes in TS2–4. To explore such interactions in vivo, we developed orthotropic PDAC mouse models with cancer cells derived from tissues of the representative TS4 (SNU3608) and IS2 (SNU3573) for TS2–4 and IS1–2, respectively. By comparing tumor growth in these models, we found that SNU3608 tumors grew faster than SNU3573 tumors. We then used fluorescence-activated cell sorting (FACS) to examine the number of tumor-infiltrating myeloid cells and found that higher levels of infiltration of PMN-MDSCs were observed in SNU3608 tumors than in SNU3573 tumors.
Subsequently, we cocultured several types of immune cells, including PMN-MDSCs, classical monocytes and nonclassical monocytes, isolated from SNU3608 tumor-bearing BALB/c nude mice with T cells from naïve BALB/c mice. We measured T cell proliferation using carboxyfluorescein succinimidyl ester (CFSE) assays. In cocultures with PMN-MDSCs, the mean fluorescence intensity of CFSE was increased for both CD8+ and CD4+ T cells compared with in control cultures, indicating that T cell proliferation was reduced as a result of exposure to PMN-MDSCs. Together with the above observations, these data suggest that PMN-MDSCs could be responsible for the depletion of anti-tumor immunity observed in TS4 with poor prognosis.
Conclusion
Our proteogenomic analysis demonstrates that proteomics data provide additional insights beyond those available with genomic data alone, and improve our understanding of PDAC. Our analysis further provides patient subtypes, subtype characteristics and potential targets for the subtypes that could be helpful in tailoring therapeutic options. Especially, based on distinct characteristics of agressive TS2-4 tumors, we proposed alternative combined therapeutic strategies for these tumors. For TS2 tumors, blockades of activated stroma, such as FAP inhibitors23, can be augmented with ROCK inhibitors24,25. For the most treatment-resistant TS4 tumors, (1) a therapy targeting PMN-MDSCs (e.g., IL10 blockers26,27) to improve CD8+ T cell-mediated anti-tumor immunity and (2) cell cycle inhibition (e.g., CDK1 inhibitors28) can be additionally considered to improve treatment efficacy. As a result, our findings can serve as a fundamental basis for making substantial advances in molecularly guided precision diagnosis and therapy of PDAC.
References
-
Collisson, E. A., Bailey, P., Chang, D. K. & Biankin, A. V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 207–220 (2019).
-
Ansari, D., Toren, W., Zhou, Q., Hu, D. & Andersson, R. Proteomic and genomic profiling of pancreatic cancer. Cell Biol. Toxicol. 35, 333–343 (2019).
-
Kafita, D., Nkhoma, P., Zulu, M. & Sinkala, M. Proteogenomic analysis of pancreatic cancer subtypes. PLoS ONE 16, e0257084 (2021).
-
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
-
Mun, D. G. et al. Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell 35, 111–124 e110 (2019).
-
Zhang, B. et al. Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 (2014).
-
Zhang, H. et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 (2016).
-
The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e13 (2017).
-
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
-
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
-
Waddell, N. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 (2015).
-
Witkiewicz, A. K. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 6, 6744 (2015).
- Vasaikar, S. et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049.e19 (2019).
-
Scarlett, C. J., Salisbury, E. L., Biankin, A. V. & Kench, J. Precursor lesions in pancreatic cancer: morphological and molecular pathology. Pathology 43, 183–200 (2011).
-
Zhang, J. et al. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database (Oxford) 2011, bar026 (2011).
-
Collisson, E. A. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503 (2011).
-
Moffitt, R. A. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178 (2015).
-
Puleo, F. et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155, 1999–2013.e3 (2018).
-
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
-
Maurer, C. et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 68, 1034–1043 (2019).
-
Peng, X. L., Moffitt, R. A., Torphy, R. J., Volmar, K. E. & Yeh, J. J. De novo compartment deconvolution and weight estimation of tumor samples using DECODER. Nat. Commun. 10, 4729 (2019).
-
Peng, J. et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 29, 725–738 (2019).
- Xin, L. et al. Fibroblast activation protein-α as a target in the bench-to-bedside diagnosis and treatment of tumors: a narrative review. Front. Oncol. 11, 648187 (2021).
- Kim, S., Kim, S. A., Han, J. & Kim, I. S. Rho-kinase as a target for cancer therapy and its immunotherapeutic potential. Int. J. Mol. Sci. https://doi.org/10.3390/ijms222312916 (2021).
-
Wei, L., Surma, M., Shi, S., Lambert-Cheatham, N. & Shi, J. Novel insights into the roles of Rho kinase in cancer. Arch. Immunol. Ther. Exp. (Warsz.) 64, 259–278 (2016).
-
Jung, K. et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Invest. 127, 3039–3051 (2017).
-
Jeong, J. et al. Tumor-infiltrating neutrophils and non-classical monocytes may be potential therapeutic targets for HER2negative gastric cancer. Immune Netw. 21, e31 (2021).
-
Wijnen, R. et al. Cyclin dependent kinase-1 (CDK-1) inhibition as a novel therapeutic strategy against pancreatic ductal adenocarcinoma (PDAC). Cancers (Basel) https://doi.org/10.3390/cancers13174389 (2021).
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in