Pushed to extremes: distinct effects of high temperature versus pressure on the structure of STEP
Published in Chemistry, Physics, and Cell & Molecular Biology
After nearly 100 years of innovation in macromolecular crystallography (MX), cryogenic X-ray data collection is now so standardized at synchrotrons that we sometimes forget the important role played by a crystal’s environment in dictating the final atomic structure. Liquid nitrogen has done wonders to prevent the rapid onset of radiation damage in protein crystals, but it effectively “locks” the protein into a fixed macromolecular conformation. However, to our benefit, data collection temperature can be easily modulated at modern synchrotrons. “Heating” crystals to room temperature and beyond during data collection has been shown to avoid structural biases found at cryogenic temperature, “flexing” the protein to reveal differences in protein conformation, ligand binding, and solvation layers.
A complementary environment that has been shown to modulate protein crystal structure is pressure. It is possible to “squeeze” a protein crystal in a high-pressure environment, achieved via high-pressure cryocooling or mounting a crystal in a diamond anvil cell. Pressurizing a protein crystal has been shown to reveal pressure-induced structural changes on the sub-angstrom level that can be directly related to protein function, conformational shifts of functional residues within allosteric networks, and changes in ligand affinity.
We know protein function hinges on small shifts of three-dimensional structure. We also know that three-dimensional structures can be flexed and squeezed using both temperature and pressure. This begs the question: why do we as structural biologists interpret structure-function relationships in cryogenic crystals as gospel, without comparing to data from crystals held under different environmental constraints?
Exploring temperature vs. pressure in crystallography
To this end, relatively few studies have explored the detailed atomic-level effects of elevated pressure on protein conformational ensembles using crystallography, while even fewer studies have compared such effects for elevated temperature vs. pressure. It thus remains unclear whether, and how, these two perturbations differentially affect protein conformational ensembles.
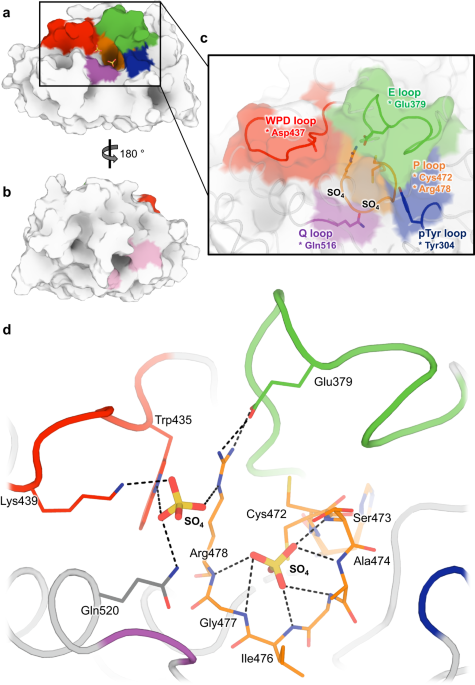
To quantitatively explore these two environmental conditions, we report the first pair of structures at physiological temperature (HiT) vs. high pressure (HiP) for the same protein and compare them to a third “reference” dataset collected at cryogenic temperature and ambient pressure (LoTP). Our target protein is STEP (PTPN5), a validated therapeutic target for Alzheimer’s disease, Fragile X syndrome, and Parkinson’s disease. Although the Protein Data Bank includes 8 crystal structures of STEP, none were collected outside of the realm of traditional cryogenic ambient-pressure crystallography.
High pressure modifies the conformational landscape of catalytic loops
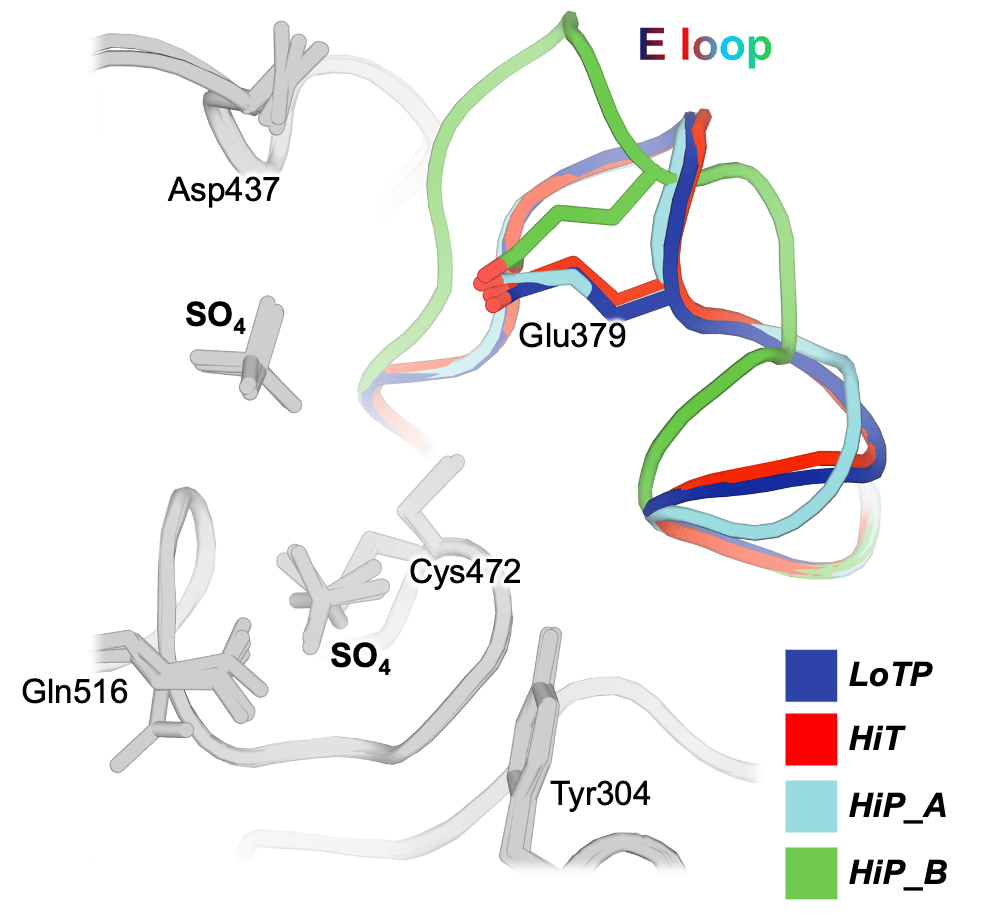
We show that these perturbations have distinct and surprising effects both locally and globally in STEP. The largest conformational changes we observe in STEP are in the E loop, a highly conserved loop that plays a critical role in regulation. Only at high pressure do we see evidence in the electron density for dual conformations of the E loop. Both conformations were individually evident in previous structures of STEP, with the “secondary” conformation previously seen only in a structure with a small-molecule allosteric activator bound elsewhere. However, our data indicates that applying pressure is sufficient to induce these conformations to coexist in a single crystal of the unliganded protein (Fig. 1). This has implications for accessing excited states of other proteins that may be relevant for function.
 and high temperature (HiT; red) but deviates into two distinct conformations at high pressure (HiP_A; cyan, HiP_B; green). Glu379, a key catalytic residue in the E loop, remains within the same conformational space regardless of E loop conformation.")
Beyond the E loop, only in the cryogenic and high-pressure datasets do we observe an ordered glycerol molecule is bound near the catalytically key Q loop. By contrast, at high temperature ordered waters are present instead, and this region of the protein undergoes conformational shifts. All three structures are from crystals treated with similar glycerol-containing cryoprotectant solutions, indicating that crystal cryocooling may induce the glycerol to bind, preventing nearby conformations with potential functional relevance seen only at physiological temperature.
Temperature and pressure alter torsional space
Globally, when embedding the entire protein structure in a reduced-dimensionality torsional space, physiological temperature shifts STEP toward previously reported active-like states, while high pressure shifts it toward a previously uncharted region. The large difference in the E loop at high pressure does not dominate this analysis, indicating that the differences between high temperature vs. pressure are driven by small, subtle conformational changes distributed throughout the tertiary structure.
Ordered water molecules are sensitive to temperature and pressure
Perturbation-induced effects are also evident for the solvation layer surrounding the protein. Our cryogenic structure has by far the most ordered water molecules observable in the electron density, high temperature has by far the fewest, and high pressure has an intermediate number. Notably, 17 (24.6%) of the HiT waters and 23 (23.5%) of the HiP waters were distinct from any LoTP water (Fig. 2). Of these 40 new positions, only 1 (2.5%) is common to both HiT and HiP. This suggests that high temperature and high pressure do not merely retain a subset of ordered waters, but rather stabilize new water positions, resulting in a distinct pattern of solvation. Such differences in constellations of water may offer useful insights into solvent energetics in ligand-binding pockets which could be exploited for rational structure-based drug design.
 All ordered water molecules at (a) LoTP, (b) HiT, and (c) HiP are shown. (d) Only the waters unique to each structure, i.e., > 2 Å from any water in the other two structures.")
Altogether, our work indicates that neither temperature nor pressure should be forgotten when analyzing the conformational ensemble of a protein. Rather, they are complementary, powerful, fundamental macromolecular perturbations – key additions to the crystallographer’s toolbox. Here’s to the next 100 years of innovation in macromolecular crystallography, with an eye toward protein structures – plural!
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in