Regulatory Transposable Elements in ENCODE
Published in Genetics & Genomics
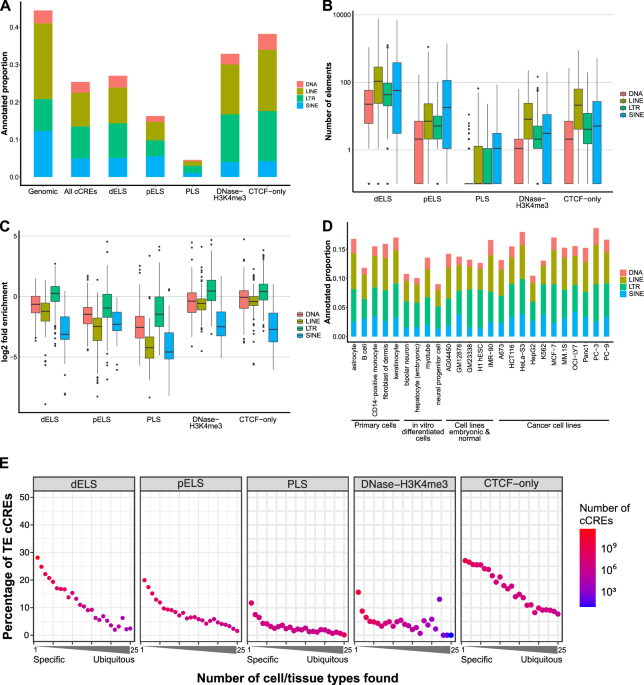
Transposable elements (TEs) are mobile genetic elements that have copied and dispersed themselves through almost all eukaryotic genomes. Compared to other mammals, humans are not particularly unusual in TE content, with ~45% of genomic sequence coming from TEs. Since TEs are a significant source of genetic material, understanding of how evolution works, especially mammalian evolution, will be incomplete until we know how TEs are involved in genome evolution.
From their discovery, it was known that TEs could affect gene expression. Generally, gene regulatory elements are sequences that determine when, where, and how much genes are expressed and likely play an important role in phenotypic variation. These elements are highly dynamic during evolution, as even closely related species can have species-specific regulatory element changes. This can make them difficult to detect by sequence conservation alone. Joining the fourth and final phase of the ENCODE consortium, we aimed to utilize the vast amount of data that has identified gene regulatory elements (candidate cis-regulatory elements, or cCREs, based on biochemical marks) to probe the relationship between TEs and regulatory element evolution.
Our first analysis looked to characterize the landscape of TE-derived cCREs in the human genome, as humans have had the most comprehensive profiling of all mammals so far. We observed that ~25% of cCREs had at least half of their sequence come from a single TE. This implies that TEs are more likely to contribute to regulatory elements than if TEs are completely neutral (0%), yet less likely than if TEs are equal to non-repetitive sequences (~45%). Also, as expected from trends in earlier studies, we saw that LTRs overrepresented in regulatory elements among TE classes. However, LINEs and SINEs were surprisingly associated with many cCREs owing to their sheer abundance.
Next, we explored how TEs can contribute to conserved and lineage-specific regulatory element evolution by comparing human and mouse cCREs. While shared cCREs were overwhelmingly identified to be in non-TE sequence, many cCREs that are only found in human or only found in mouse come from TEs. During this analysis, we noticed something interesting but slightly confusing: TEs in human would sometimes be assigned to a different, but related subfamily in mouse even though the TE is found in the same relative genomic location. In other words, the sequence synteny suggests that the TEs should be the same, but the annotation was incorrect. We realized that uncertainty in assigning a TE to a specific subfamily increases with age and more closely related subfamilies. Thus, the challenge of doing this correctly (or at least consistently) becomes increasingly more difficult with more species, each with its own set of lineage-specific mutations. Similarly, TEs past several hundred million years become almost impossible to detect apart from very well preserved TEs. These practical limitations in TE annotation, while very technical, argue that what we observe for TE contributions to regulatory elements may be a lower bound.
Gene regulatory elements are thought to exert their effects through the binding and activity of transcription factors (TFs). Many groups, including us, had previously found that different TF binding sites (TFBSs) are enriched in different TE subfamilies. In some cases, TFBSs look to have originated from the ancestral source TE that gave rise to the entire subfamily, yet in others, TFBSs appear to have arisen through mutations in a sub-lineage. We were curious if TFBS that could be involved in gene regulatory activity are frequently coming from ancestral sequence in general. To identify potential regulatory TFBS, we searched for TFBS motifs that are enriched in cCRE associated TEs for each subfamily. Then, we inferred an ancestral origin for motifs for each individual TE copy based on alignment to the TE subfamily consensus sequence (representing the ancestral state). Interestingly, LINE, LTR, and DNA transposon TE classes show evidence that their TFBS motifs are more likely to be derived from an ancestral motif than expected by chance. On the other hand, SINEs appear to mostly have non-ancestral motifs, likely created through random mutations instead. These results suggest that SINEs may generally evolve regulatory activity in a fundamentally different manner compared to other TE classes.
TEs typically create new insertions relatively randomly in the genome. Based on previous research, we hypothesized that insertion location may influence whether a specific TE copy becomes a regulatory element. We found that TEs associated with cCREs or TFBSs are relatively close to non-TE cCREs or TFBSs, suggesting that there is a location effect. We were unable to further explore potential mechanisms for this phenomenon, but we believe this is a fascinating area of research to follow up.
While most focus in the field has been on TEs providing novel functions, TEs may also allow for greater “evolutionary flexibility” by contributing redundant functional sites. Functional redundancy can relax purifying selection to potentially allow for adaptation of new functions, separation of functions for further specialization or specificity, or simply decay by inactivating mutations. We investigated cases of observable changes in TFBS between human and mouse in analogous cell types to estimate TE contribution to turnover, where a TE provides TFBS that replaces the original. From our identified putative TFBS turnover sites, we found that 3-56% occur at TEs depending on the TF, suggesting that TEs could be a major contributor for maintenance of TFBS during evolution. It would be interesting to see future studies expand on the concept of turnover of functional elements and how TEs factor into the equation.
Lastly, we asked the question of whether the sequence origin of a regulatory element, that is whether the underlying sequence is TE or non-TE derived, can distinguish types of regulatory elements. One of the new experiment types in ENCODE4 is massively parallel reporter assay (MPRA), which functionally tests sequences for regulatory activity. We found that sequences that are mostly TE-derived display equal if not higher activity in MPRA compared to non-TE sequences for the over 100,000 total tested sequences. Interestingly, this was consistent with a previous study where we focused on a single TE subfamily that appeared have very high regulatory activity (even compared to our positive controls) and gradually lost that activity over time before some copies were potentially co-opted as regulatory elements. Furthermore, TE-derived cCREs were similar to non-TE cCREs based on MPRA, ATAC-seq for open chromatin, phastCons score for sequence conservation, and TF binding. In the human population, TE-derived cCREs are slightly depleted for common variants and enriched for GWAS associated variants much like their non-TE counterparts, suggesting that these TE regulatory sequences are important for health and disease.
Altogether, our study provides systematic analyses to more generally describe how TEs have contributed to the regulatory genome. As the evolutionary history of most eukaryotic genomes is inescapably linked to the TEs that inhabit them, we hope that this work sparks interest in further understanding how TEs can start as “junk DNA” but end up providing various useful functions.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in