Rescuing retromer in Parkinson's disease
Published in Neuroscience and Genetics & Genomics
Why we do the research?
Parkinson’s disease (PD) is the most common age-associated movement disorder that can be directly and unequivocally attributed to the loss of a population of dopamine neurons. PD is also associated with a constellation of disabling autonomic, cognitive, psychiatric, sensory and sleep problems, all of which are poorly responsive to medication. While dopamine-replacement therapies and deep brain stimulation may relieve motor symptoms, they do not slow or stop disease progression. We reason that to predict and prevent PD, research must first identify its molecular components and understand how they operate. Secondarily, research must model and test how that molecular dysfunction leads the selective vulnerability and loss of dopaminergic neurons. On this premise, we anticipate clinical trials of novel disease interventions will have the highest likelihood of success.
We have begun to elucidate the genetic etiology of PD. Our team performed the linkage and molecular genomic analyses that lead to the original discoveries of pathogenic variants in leucine-rich repeat kinase 2 (most notably LRRK2 p.G2019S) (Zimprich A. et al., 2004; Kachergus J. et al., 2005) and vacuolar protein sorting 35 (VPS35 p.D620N) (Vilariño-Güell C. et al., 2011). Many individuals in these families received a clinical diagnosis of typical late-onset PD, and nearly half of the post-mortem brains examined are confirmed to have Lewy body disease. However, there are considerable differences in clinical and pathologic presentations, even among close relatives, and the probability of the phenotype given the mutant genotype is not absolute. Idiopathic PD is multifactorial, and while genetics loads the gun, environment is likely to be the trigger. Nevertheless, these Mendelian forms of parkinsonism are dominantly-inherited and their effect size far exceeds any locus implicated by association studies of sporadic PD. Such discoveries justify and enable functional studies in genetically-modified mice (Hinkle K. et al., 2012, Yue M. et al., 2015, Cataldi S. et al., 2018). These are models of gene dysfunction; it is naïve to expect them to develop PD, although being faithful to the human cause they may recapitulate important aspects of disease pathogenesis.
The problem of pleiotrophy
Many prior studies demonstrate pathogenic variants in leucine-rich repeat kinase 2 (LRRK2) activate its kinase activity, and none more directly than p.G2019S that keeps the hinge of its ‘activation segment’ ajar. However, LRRK2 is a large multi-domain protein and has many substrates, most notably a set of Rab GTPases (Steger M. et al., 2016). In brain, LRRK2 also phosphorylates many synaptic components with subtle albeit significant impacts on dopaminergic and glutaminergic neurotransmission (expertly reviewed by Kuhlmann & Milnerwood, 2020; Pischedda & Piccoli 2021). Nevertheless, LRRK2 is most highly expressed in neutrophils, monocytes and T-cells, so studies in the immune system are in vogue. Indeed, LRRK2’s direct functional relevance to dopamine neurons has long been contested, given its low level of expression. In contrast, VPS35 is a core component of the retromer complex with ubiquitous but precisely balanced expression. The retromer is responsible for recycling many thousands of different membrane-associated protein cargos between different membrane compartments, that vary by cell type. The biological complexity this presents is as fascinating as it is vast. However, we are driven to ask “What aspect of this biology is most relevant to the pathophysiology of PD?”
Making challenges opportunities
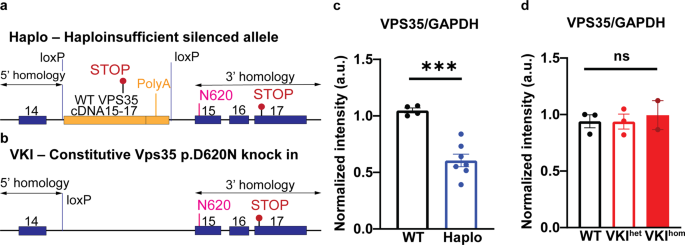
To tease out cell-specific expression we designed a conditional mutant Vps35 p.D620N model to produce wild type protein (a chimera of Vps35 exons 15-17 cDNA fused to a bovine growth hormone poly-A 3' UTR), until that 'floxed' mini-gene was ablated. As the mini-gene was inserted into intron 14 we introduced the p.D620N substitution downstream into endogenous exon 15. Experimentally, we hoped to target dopaminergic neurons in vivo by expressing cre recombinase under a partial tyrosine hydroxylase promoter (AAV2 Th:cre), so that wild type cells could be made mutant. However, with the same design, we might use germline cre to develop a constitutive Vps35 p.D620N knock-in model. While the latter was successful, the presence of the mini-gene cassette silenced its recombinant allele. We assumed the endogenous 3' UTR controls the strict 1:1:1 stoichiometry of the retromer core trimer (Vps26, Vps29, Vps35). Ozgene, our commercial partner tried again (at their cost) with an improved construct, but that also resulted in allele silencing, and why remains enigmatic. Hence, this study compared Vps35 p.D620N knock-in mice (VKI) with haplo-insufficient animals that have 50% less Vps35 expression (Haplo). Given both lines originated from the same founder we thought to explore Vps35 p.D620N gain- versus loss-of-function. Importantly, we questioned "Does VPS35 pD620N constitutively activate LRRK2 kinase because of a general deficit in retromer function or is that specific to the p.D620N mutation?”
We previously published Vps35 p.D620N mice and described differences in dopamine physiology measured using fast-scan cyclic voltammetry in electrically-stimulated, striatal brain slices (Cataldi S. et al., 2018). The signal reflects dopamine in the tissue, its release and reuptake, and VKI slices generate almost twice the amplitude of response compared to wild type. In these mice direct measurement of dopamine and its metabolites by striatal microdialysis also showed the ratio of dopamine metabolites (HVA and DOPAC)/dopamine was significantly elevated. In addition, Western blotting showed a decrease in striatal DAT and increase in VMAT2. Despite these differences nigrostriatal morphology at 3 months of age was preserved, and the animals had no obvious movement disorder. However, to interpret neuroscience without behavior is analogous to understanding genetics without evolution. Thus, we decided to pharmacologically ‘poke a stick at it’ using amphetamine to see if we might evoke behavioral differences.
We appreciate LRRK2 and VPS35 directly interact as each can be immuno-precipitated by pulling down the other. In 2017, we also learned DAT was a retromer cargo (Wu S. et al., 2017). Hence, we reasoned DAT might provide a quantitative and cell-specific marker for dopaminergic terminals within the striatum, and for the VPS35 and LRRK2 biology within them. We have previously imaged profound changes in 18F-DOPA turnover, methylphenidate- (a surrogate for DAT) and tetrabenazine-binding (a surrogate for VMAT2) in affected and asymptomatic LRRK2 p.G2019S heterozygotes (Sossi V. et al., 2010; Wile J. et al., 2017). Thus, we wondered “Would VKI mice, and perhaps human VPS35 p.D620N heterozygotes, benefit from LRRK2 kinase inhibitors?”
Interpreting results and moving forward
In Bu M. et al., 2023 we put this biology together, and remarkably invoked a pharmacological rescue of several of the dopaminergic phenotypes observed. In brief, we show how Vps35 p.D620N compromises dopamine physiology in vivo, and how that deficit leads to related amphetamine-induced hyper-locomotion. Furthermore, we demonstrate how that aberrant behavior and pathophysiology can be rescued by the administration of LRRK2 kinase inhibitors. While we focus on DAT recyling and autophagy, inhibition of LRRK2 kinase also releases the ‘brake’ on striatal output reducing amphetamine-induced movement. These results and this model may inform and accelerate pre-clinical development of LRRK2 kinase inhibitors, to slow/halt disease progression and speed their entry into clinical trials.
To most with PD, or to those with a genetic propensity to develop it, neuroprotection is an unmet need, so the results in Bu M. et al., 2023 are a major win. Mouse models of Vps35 p.D620N gene dysfunction now provide a platform to compare LRRK2 inhibitors. Non-invasive human imaging studies in VPS35 p.D620N heterozygotes may similarly inform target engagement and functional efficacy, perhaps longer-term and for the lowest dose. Biogen has already licensed LRRK2 kinase inhibitors for almost US $1 billion with the hope of bringing them to market. Pragmatic investments in the etiology and ontology of PD may enable that disease-modification (neuroprotection) to become more than a noble ambition.
Follow the Topic
-
npj Parkinson's Disease
This journal publishes original basic science, translational and clinical research related to Parkinson's disease, including anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology and therapeutic development and treatments.

Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcementRelated Collections
With Collections, you can get published faster and increase your visibility.
The neuroimmune-axis and ageing in Parkinson’s Disease
Publishing Model: Open Access
Deadline: Jul 15, 2026
Cognition - preclinical models, and preclinical unmet need
Publishing Model: Open Access
Deadline: Jul 27, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in