SARS-CoV-2 virulence factor ORF3a blocks lysosome function by modulating TBC1D5-dependent Rab7 GTPase cycle
Published in Microbiology and Cell & Molecular Biology
The Pandemic
The worldwide pandemic of Coronavirus Disease 2019 (COVID-19) caused by the novel Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) has overwhelmed hospitals and claimed millions of human lives. This pandemic continues to cause a devastating health crisis throughout the world, with more than 7 million deaths by the year 2024. The fatality rate of COVID-19 is very high when compared to other virus infections. Adaptive mutation in SARS-CoV-2 has led to the emergence of multiple natural variants with different characteristics in comparison to its wild-type strain. Considering their potential effects on public health worldwide, some of these variants have been declared variants of concern (VOCs). The emergence of vaccines undoubtedly changed the course of the pandemic globally.
Background
SARS-CoV-2 is classified as a beta-coronavirus (β-CoV) with a positive sense single-stranded RNA. The 5' genomic region of SARS-CoV-2 makes up more than two-thirds of the genome and encodes orf1ab polyproteins, whereas the 3' region contains genes encoding structural proteins such as spike (S), envelope (E), membrane (M), and nucleocapsid (N). Furthermore, six accessory proteins encoded by ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10 are present in the 3’ region of the virus genome.
Conventionally, major roles played by viral accessory proteins include subverting and/or disrupting host cellular pathways, escaping immune evasion, and increasing infectivity. Many pathogens, from bacteria to viruses, modulate the vesicular transport system of the host to aid in their growth and survival. To this end, there is a growing interest in understanding the mechanisms by which effectors or virulence factors of pathogens disrupt the intracellular transport system.
Among the SARS-CoV-2 accessory proteins, the largest protein encoded is ORF3a. Interestingly, the SARS-CoV-2 ORF3a is approximately 90% similar and 72% identical in nucleotide sequence with the ORF3a of the SARS-CoV-1. Structurally, the SARS-CoV-2 ORF3a protein contains three transmembrane (TM) helices and a cytosolic domain. Previous studies have shown that SARS-CoV-2 ORF3a interacts directly with Vps39, a subunit of the HOPS complex. HOPS is a heterohexameric complex that localizes to lysosomes and mediates tethering and SNARE-dependent fusion of lysosomes with other membrane-bound compartments such as late endosomes and autophagosomes1,2 3. ORF3a disrupts interaction between subunits of the HOPS complex and with late endosomal GTPase Rab7. ORF3a impairs the assembly of the trans-SNARE complex required for the fusion of autophagosomes with lysosomes1,2. Also, another study revealed that ORF3a promotes lysosome exocytosis by facilitating recruitment of BORC complex and Arl8b, and ectopic expression of ORF3a in the coronavirus MHV-A59 (which lacks ORF3a) promotes viral egress outside the host cell3. Indeed, β-CoV has been shown to egress from the host cell after infection through the Arl8b-positive lysosomal-dependent pathway4.
Exciting Results!!!
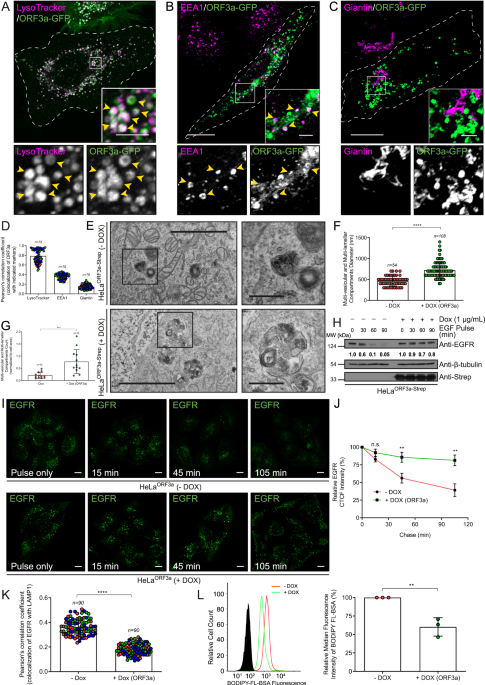
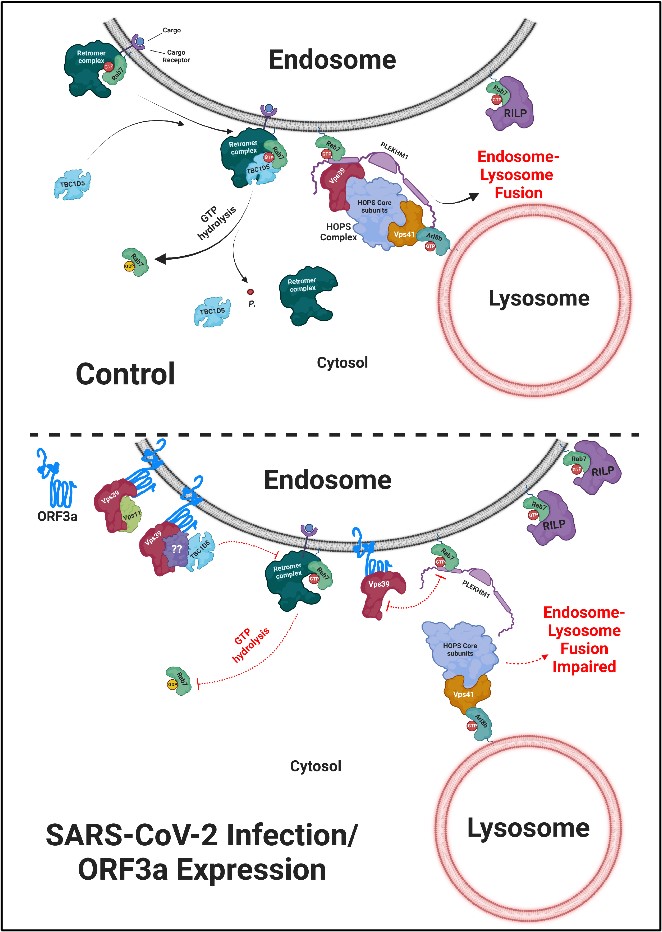
In this study, we found that the localization of ORF3a to lysosomes is crucial for the disruption of endocytic as well as autophagic degradative pathways. Previous studies have shown that PLEKHM1, a Rab7 effector, interacts directly with Vps39 and is responsible for its recruitment on the late endosome membranes. This Vps39-PLEKHM1 complex is crucial for the assembly of the complete tethering factor HOPS complex and further facilitates the recruitment of SNARE proteins to mediate the fusion of the autophagosome/late endosome with the lysosome5,6. We questioned whether the presence of ORF3a affects the interaction between PLEKHM1 and Vps39 and found that ORF3a abrogates Vps39 interaction with PLEKHM1. Our results indicate that by disrupting the interaction between Vps39 and PLEKHM1, ORF3a blocks endocytic and autophagic cargo degradation in lysosomes.
Interestingly, we found increased endosomal localization of PLEKHM1 in ORF3a-expressing cells. Since PLEKHM1 is an effector of Rab7, we investigated the activation status of Rab7 and found that expression of ORF3a or SARS-CoV-2 infection leads to a dramatic increase in active or GTP-bound Rab7 levels. We found that ORF3a expression and Rab7 activation were essential for SARS-CoV-2 replication, indicating a key role of ORF3a-mediated Rab7 activation as an essential step during viral pathogenesis. At least one of the consequences of ORF3a-mediated active Rab7 was a defect in recycling of the cation-independent mannose-6-phosphate receptor (CI-M6PR) from Rab7-positive endosomes to the trans-Golgi network (TGN). CI-M6PR binds and sorts newly synthesized mannose-6-phosphate-conjugated cathepsins (lysosomal hydrolases) from the TGN to the late endosomes. In line with the observed defect in CI-M6PR recycling, we found impaired cathepsin processing in lysosomes and increased procathepsin secretion in ORF3a-expressing cells. ORF3a expression and SARS-CoV-2 infection also impaired the fusion and maturation of Rab7-positive compartments into Arl8b and LAMP1-positive compartments. It is to be noted that instead of fusing with late endosomes, Arl8b and LAMP1-positive vesicles likely serve as vehicles for virus egress from the infected host cells. To understand the mechanism by which ORF3a mediates Rab7 activation, we assessed whether ORF3a affects the function of Rab7 GAPs. We found that ORF3a promotes the formation of an ORF3a-Vps39-TBC1D5 complex, wherein TBC1D5 is a known GAP of Rab7 on late endosomes. Concurrently, the interaction of Rab7 with its GAP TBC1D5 was greatly reduced in ORF3a-expressing cells. We confirmed our findings in SARS-CoV-2-infected cells and found that ORF3a expression and its interaction with Vps39 are crucial for Rab7 hyperactivation in infected cells. Taken together, our results suggest that SARS-CoV-2, through its accessory protein ORF3a, inhibits both endocytic as well as autophagic degradative pathways by disrupting the Vps39-PLEKHM1 complex. Furthermore, ORF3a stalls GTP hydrolysis of Rab7, which impairs the transport of newly synthesized hydrolases to lysosomes and the fusion of lysosomes with other compartments, consequently promoting the egress of the virus via lysosomal exocytosis.
Conclusions
Taken together, the findings imply that during SARS-CoV-2 infection, ORF3a stalls the GTPase cycling of Rab7, an indispensable regulator of late endosomes, as well as lysosomal positioning and function. ORF3a impairs the fusion of late endosomes with lysosomes, but rather promotes the virus's egress via lysosomal exocytosis and supports the viral infection inside the host.
Link to the study:
https://www.nature.com/articles/s41467-024-46417-2
References:
1 Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell 56, 427-442 e425, doi:10.1016/j.devcel.2020.12.010 (2021).
2 Zhang, Y. et al. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov 7, 31, doi:10.1038/s41421-021-00268-z (2021).
3 Chen, D. et al. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell 56, 3250-3263 e3255, doi:10.1016/j.devcel.2021.10.006 (2021).
4 Ghosh, S. et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 183, 1520-1535 e1514, doi:10.1016/j.cell.2020.10.039 (2020).
5 McEwan, D. G. et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57, 39-54, doi:10.1016/j.molcel.2014.11.006 (2015).
6 Marwaha, R. et al. The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J Cell Biol 216, 1051-1070, doi:10.1083/jcb.201607085 (2017).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in