SignalP 5.0 improves signal peptide predictions using deep neural networks
Published in Bioengineering & Biotechnology

When I started working with signal peptide (SP) prediction as a biology Master student in 1991, I had no idea how important the task was to the biochemical community. It was just one of several ideas that my then supervisors at the Technical University of Denmark, Søren Brunak and Jacob Engelbrecht, had in their pipeline. I found it interesting and imagined that it would be possible to finish within the scope of an M.Sc. project.
This turned out not to be the case. Those were the early days of bioinformatics, and no textbooks had been written yet, so many approaches had to be invented along the way. Among these was the criteria for homology reduction of the data set, which we investigated in a publication from 1996 [1]. In the same year, the first version of our artificial neural network (ANN) based method SignalP was put online as a web server [2], and I soon experienced an overwhelming interest in the program from researchers around the world — an interest that prompted me to stay in the field. By then, I had become a PhD student under the supervision of Gunnar von Heijne of Stockholm University and Søren Brunak.
SignalP version 1 was in 1998 followed by version 2 which included a hidden Markov model (HMM) alongside the ANNs [3]. SignalP 2 was to some degree able to discriminate between SPs and signal anchors (uncleaved transmembrane segments close to the N-termini of the proteins). SignalP 3 from 2004 featured retrained versions of both the ANN and HMM parts, and introduced a new score for discriminating between SPs and other proteins [4]. In 2011, SignalP 4 showed a drastically improved discrimination between SPs and transmembrane segments using only ANNs [5].
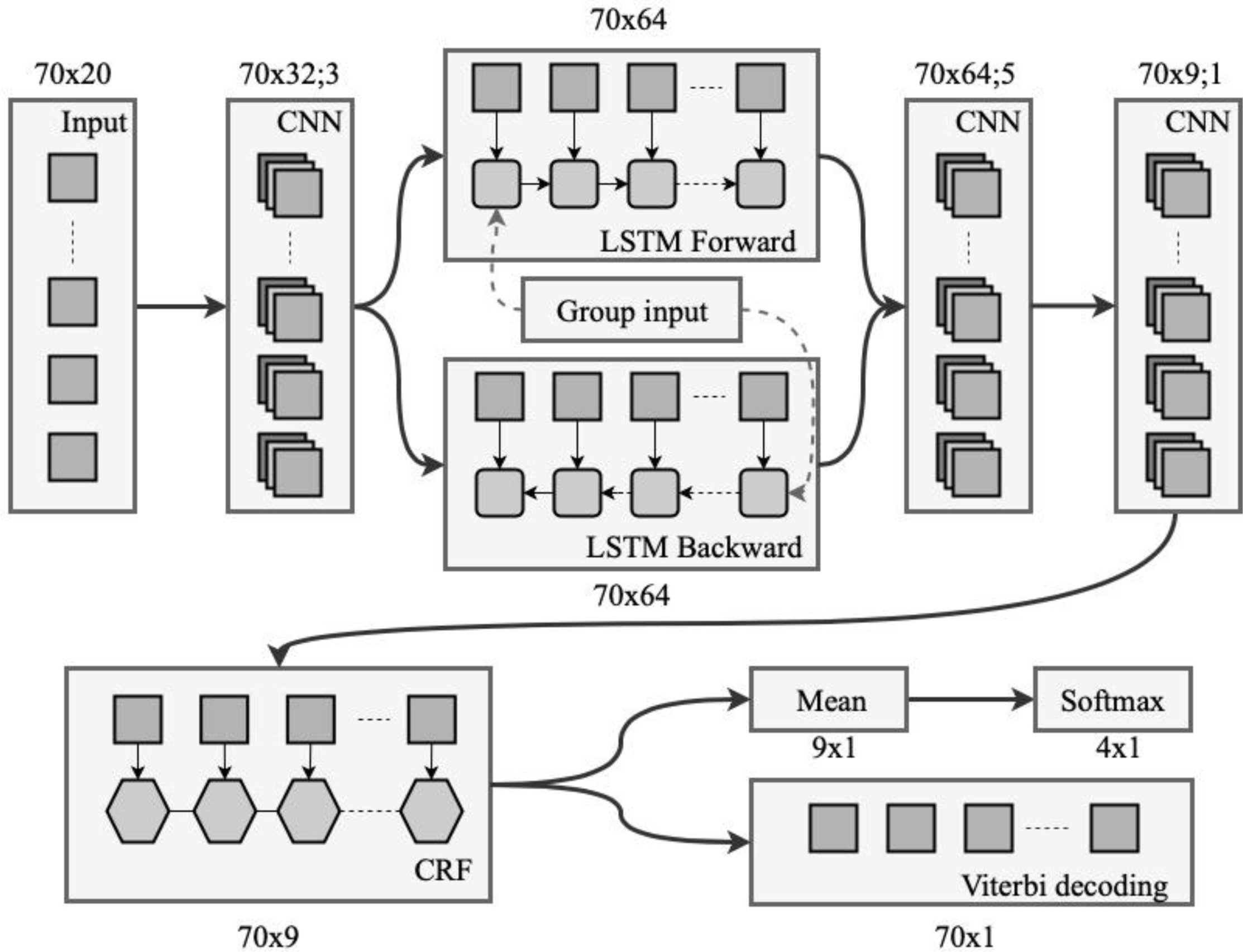
Now, we introduce SignalP 5.0 which is based on deep learning. The deep recurrent ANN architecture is better suited to recognizing sequence motifs of varying length, such as SPs, than traditional feed-forward ANNs (as used in SignalP 1-4). The ANNs in SignalP 5.0 are further combined with a conditional random field (CRF), which imposes a defined grammar on the prediction and obviates the need for the post-processing step used in earlier versions of SignalP. The architecture of SignalP 5.0 is shown below.
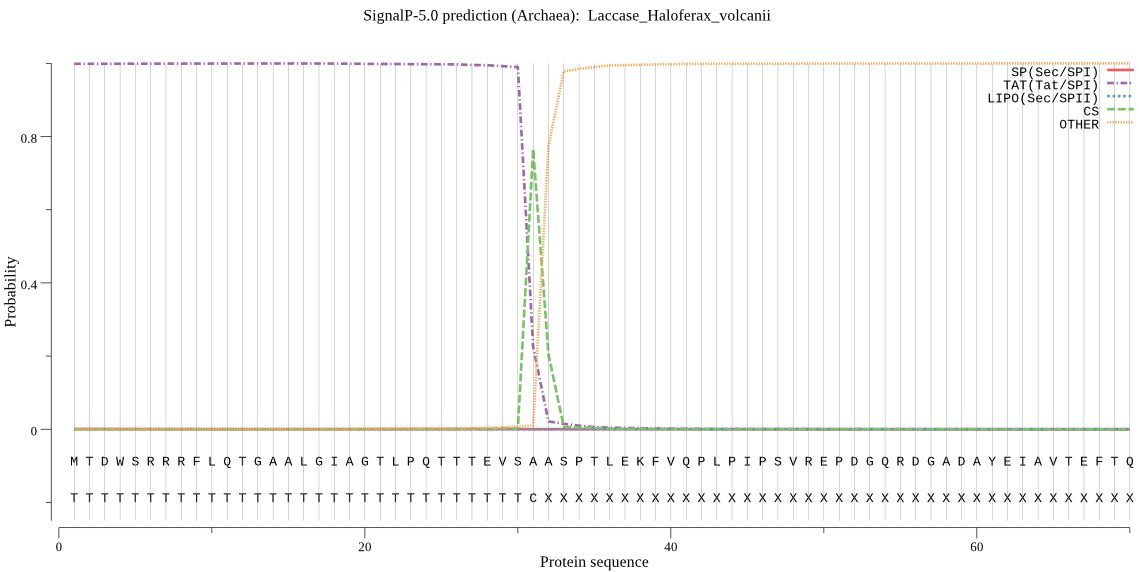
SPs from different organisms share an overall structure, but there are differences between Gram-negative bacteria, Gram-positive Bacteria, Archaea and Eukarya. Therefore, SignalP 1-4 divided the data set into systematic groups that were trained and tested separately. SignalP 5.0 instead exploits the shared overall structure and is trained on data from all groups together, while an extra input unit informs the network about the origin of the sequences. In this way, we have been able to make predictions also of SPs from the previously ignored Archaea, even though the data set for this group is severely limited.
Another novelty is that SignalP 5.0 can differentiate between “standard” signal peptidase I-cleaved SPs translocated by the Sec translocon (Sec/SPI) and two other types of SPs in prokaryotes, namely lipoprotein SPs cleaved by signal peptidase II (Sec/SPII) and "Tat" (Twin-Arginine Translocation) SPs translocated by the Tat translocon (Tat/SPI). Previously, we referred to two separate servers, LipoP [6] and TatP [7] respectively, for such predictions. However, SignalP 5.0 cannot predict lipoprotein SPs translocated by the Tat translocon (Tat/SPII) since we did not find any confirmed examples of these while constructing the data sets.
In our benchmarks, SignalP 5.0 compares favorably with other SP prediction methods in almost all cases. The exceptions are mainly cleavage site prediction in Archaea and Tat SP prediction in Bacteria, where the specialized predictors PRED-SIGNAL [8] and PRED-TAT [9], respectively, did better on some measures. However, there was an overlap between our benchmark data set and the data sets of those specialized predictors, implying that their performances are not true test values, while those of SignalP 5.0 are.
So, have I then come to the end of my personal SP prediction road that began in 1991? Probably not. Besides the above-mentioned question of the Tat/SPII sequences, SignalP 5.0 is also not able to predict the special SPs of Type IV pilins, which are cleaved by signal peptidase III (a.k.a. prepilin peptidase). In addition, the possible differences between SPs of various bacterial phyla or eukaryotic kingdoms have not been fully explored. There is still work to do!
1. Nielsen H, Engelbrecht J, von Heijne G, Brunak S. Defining a similarity threshold for a functional protein sequence pattern: The signal peptide cleavage site. Proteins Struct Funct Bioinforma. 1996;24:165–77.
2. Nielsen H, Brunak S, Engelbrecht J, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng Des Sel. 1997;10:1–6.
3. Nielsen H, Krogh A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc Int Conf Intell Syst Mol Biol. 1998;6:122–30.
4. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95.
5. Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Meth. 2011;8:785–6.
6. Juncker AS, Willenbrock H, von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–62.
7. Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinformatics. 2005;6:167.
8. Bagos PG, Tsirigos KD, Plessas SK, Liakopoulos TD, Hamodrakas SJ. Prediction of signal peptides in archaea. Protein Eng Des Sel. 2009;22:27–35.
9. Bagos PG, Nikolaou EP, Liakopoulos TD, Tsirigos KD. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics. 2010;26:2811–7.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in