SignalP 6.0 predicts all five types of signal peptides using protein language models
Published in Bioengineering & Biotechnology
Explore the Research
SignalP 6.0 predicts all five types of signal peptides using protein language models - Nature Biotechnology
A new version of SignalP predicts all types of signal peptides.
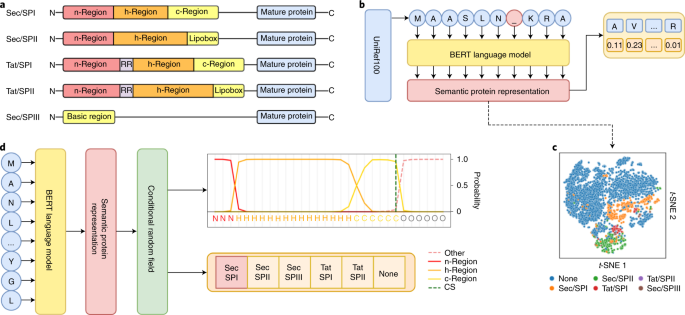
It is well known that signal peptides (SPs) occur in both eukaryotes and prokaryotes. A less well known fact is that there are several types of SPs in prokaryotes (both Bacteria and Archaea). There are two types of translocons (protein translocation channels) and three types of signal peptidases (the enzymes responsible for cleaving the SP from the rest of the protein).
One of the best known methods for predicting SPs from amino acid sequences is our program SignalP, which has been online since 1996. A very brief history of SignalP can be found in the Behind the Paper post “SignalP 5.0 improves signal peptide predictions using deep neural networks” from February 2019. A more elaborate history of SignalP and related programs can be found in the open access paper “A Brief History of Protein Sorting Prediction” from the same year [1].
The quest for complete signal peptide prediction
When we wrote the SignalP 5.0 manuscript [2], we were convinced that we had made an SP prediction method that was as complete as possible — in addition to the “standard” SPs transported by the Sec translocon (SecYEG) and cleaved by signal peptidase I (also known as leader peptidase, Lep), SignalP 5.0 could predict two other types:
- Tat SPs transported by the alternative Tat translocon (TatABC), and
- lipoprotein SPs cleaved and lipid-modified by lipoprotein signal peptidase, also known as signal peptidase II (Lsp).
Thereby, SignalP 5.0 was supposed to make our two older special-purpose prediction methods TatP and LipoP obsolete.
However, the reviewers assigned by Nature Biotechnology pointed out that this was not enough to make SignalP 5.0 complete. One remaining issue was the special short SPs of type IV pilins and related proteins, cleaved by a third signal peptidase known as prepilin peptidase (PilD) or signal peptidase III (sometimes even called signal peptidase IV). We were aware that these SPs existed, but found too few examples in the databases to make deep learning for this type feasible.
The other remaining issue was, admittedly, a surprise to us: Some proteins are simultaneously “Tat” and “lipo”, i.e. they are transported by the Tat translocon and cleaved and lipid-modified by signal peptidase II. SignalP 5.0 could not predict these, as it had two separate, mutually exclusive, branches for Tat proteins and lipoproteins. When we checked the databases (UniProt and PROSITE), we failed to find even a single example of this group of proteins, yet there is evidence in the literature for their existence [3,4].

We discovered a reason for the absence of Tat/SPII examples from PROSITE: the “Twin arginine translocation (Tat) signal” profile, PS51318, has a post-processing rule that excludes the “Prokaryotic membrane lipoprotein lipid attachment site” profile, PS51257. However, by manually modifying the PS51318 entry, we were able to create a small set of proteins that matched both of these profiles.

Protein language models
Now the question was: how to create a deep learning method that could learn to recognize Tat-lipo and type IV pilin-like SPs, when the positive datasets had only 38 and 113 sequences (before partitioning), respectively? The answer came from a seemingly completely unrelated field: language modeling.
Language models (LMs) are used in Natural Language Processing and are made by training an artificial neural network to predict words that have been masked out of sentences written in a human language. Recently, the idea arose that protein LMs could be created in an analogous way by masking out one amino acid at a time in naturally occurring protein sequences and asking an artificial neural network to predict the missing amino acid from its context [5-7]. The point is that a trained protein LM has learned some general features of proteins that can help predict specific protein properties, referred to as downstream tasks. Since the training of the LM does not require any labels or annotations, it can be carried out using a huge database with millions of sequences, while the downstream tasks are done with smaller labeled datasets.
Previous results have shown that protein LMs trained on millions of sequences can learn to generalize from very few examples [8]. Therefore, we hypothesized that a protein LM would be able to learn the characteristics of the rare types of SPs, Tat/SPII and Sec/SPIII.
We used a pretrained protein LM taken from the project ProtTrans [9]. In order to perform better in our downstream task of SP prediction, we fine-tuned the model. Fine-tuning improved the separation of the various categories of SPs and non-SPs, but as can be seen from the figure below, the ProtTrans model “knew” something about SPs already before fine-tuning.

Results
Indeed, our results show that SignalP 6.0 is able to learn the rare SP types. As can be seen in panel a of the figure below, SignalP 6.0 is better at recognizing almost all categories than a version of the SignalP 5.0 architecture retrained on our new dataset. The improvement is especially drastic for the rare types, Tat/SPII and Sec/SPIII, where the SignalP 5.0 performance is generally too low to be practically useful. Furthermore, the advantage of SignalP 6.0 over the SignalP 5.0 architecture is higher for sequences that are distantly related to those in the training set (panel c in the figure below), underlining the excellent generalization ability of the protein LM.

We also found that SignalP 6.0 does not “care” about the taxonomic origin of the sequences. The model was informed during training whether each sequence originated from Eukarya, Gram-positive bacteria, Gram-negative bacteria, or Archaea. We carried out an experiment shuffling the organism group labels and testing the models again, and while that operation substantially deteriorated the SignalP 5.0 performance, it did not affect SignalP 6.0 performance (see figure below). This carries two advantages: First, SignalP 6.0 can be used for sequences where the origin is unknown, such as metagenomic datasets; second, it is directly applicable to bacteria that are ambiguous with respect to the Gram-positive/Gram-negative divide, such as Thermotogae, Tenericutes, or Negativicutes. The final SignalP 6.0 server just has an option for selecting “Eukarya” or “Other”, where the only difference is that the “Eukarya” option suppresses predictions of other types than Sec/SPI (the only type of SP in the ER membrane).

Signal peptide regions
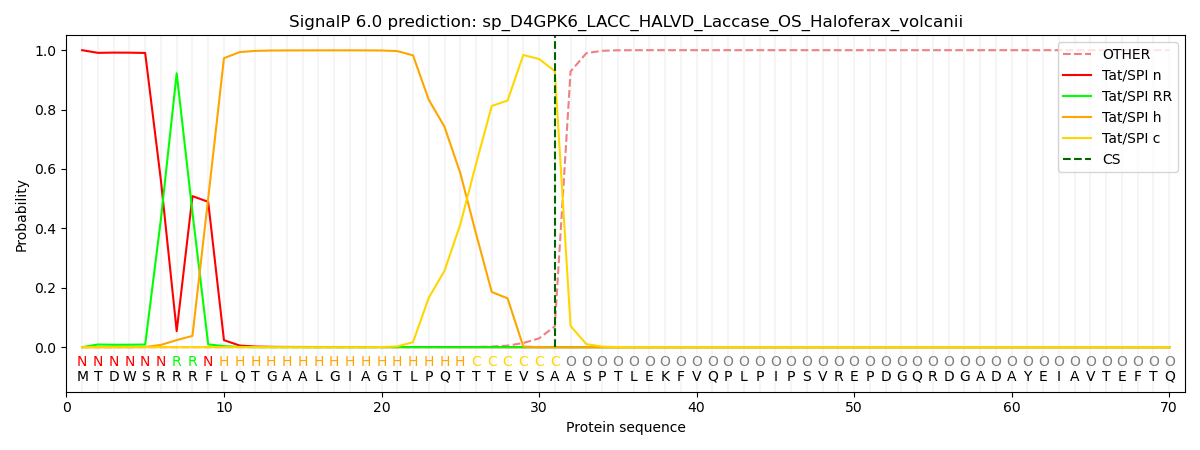
An additional feature of SignalP 6.0 is the assignment of region borders. As shown in the figure above, SPs are generally described as consisting of 2-3 regions (except for the short Sec/SPIII sequences). While assignment of the regions in a particular SP sequence previously required the trained eye of an expert, SignalP can now do it automatically. Since there is no “gold standard” here (experts may disagree on the exact region borders), we trained this task using semi-supervised learning where only a few positions had unambiguous labels: In Sec/SPI SPs, the first two positions were n-region, the last three positions were c-region, and the central position in the most hydrophobic 7-residue window was h-region. The borders were then found by maximizing the difference in amino acid composition between the regions.
Who needs region assignments? One advantage is that they can be used to characterize SPs from various organism groups, another is that they are relevant for biotechnological applications. One study from 2020 created a library of synthetic SPs and tested their ability to secrete proteins in Bacillus subtilis [10]. The authors found no systematic differences between the functional and the non-functional SPs, but after assigning regions, we could show that the functional SPs had significantly more charged and less hydrophobic n-regions than the non-functional examples (see Supplementary Figure 6 in the paper).
References
- Nielsen H, Tsirigos KD, Brunak S, von Heijne G (2019) A Brief History of Protein Sorting Prediction. Protein J 38:200–216. https://doi.org/10.1007/s10930-019-09838-3
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, et al (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology 37:420–423. https://doi.org/10.1038/s41587-019-0036-z
- Storf S, Pfeiffer F, Dilks K, et al (2010) Mutational and Bioinformatic Analysis of Haloarchaeal Lipobox-Containing Proteins. Archaea 2010:410975. https://doi.org/10.1155/2010/410975
- Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC (2009) Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold ‘em, knowing when to fold ‘em. Trends in Microbiology 17:13–21. https://doi.org/10.1016/j.tim.2008.10.001
- Alley EC, Khimulya G, Biswas S, et al (2019) Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16:1315–1322. https://doi.org/10.1038/s41592-019-0598-1
- Heinzinger M, Elnaggar A, Wang Y, et al (2019) Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics 20:723. https://doi.org/10.1186/s12859-019-3220-8
- Rives A, Meier J, Sercu T, et al (2021) Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. PNAS 118:e2016239118. https://doi.org/10.1073/pnas.2016239118
- Biswas S, Khimulya G, Alley EC, et al (2021) Low-N protein engineering with data-efficient deep learning. Nat Methods 18:389–396. https://doi.org/10.1038/s41592-021-01100-y
- Elnaggar A, Heinzinger M, Dallago C, et al (2021) ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing. In IEEE Transactions on Pattern Analysis and Machine Intelligence, forthcoming. https://doi.org/10.1109/TPAMI.2021.3095381
- Wu Z, Yang KK, Liszka MJ, et al (2020) Signal Peptides Generated by Attention-Based Neural Networks. ACS Synth Biol 9:2154–2161. https://doi.org/10.1021/acssynbio.0c00219
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in