How do electron transfer reactions work at a molecular level? Researchers have been seeking the answer to this question for decades. In Marcus theory of electron transfer, the potential energy surfaces of reactants and products are drawn as a function of a single solvent coordinate. While this exemplifies the crucial role of the solvent environment, we are left to wonder what are the specific solvent motions that control (and that we can use to control) electron transfer reactions. Is it translation or rotation of the solvent molecules? Is it a global reorganization in response to the charge redistribution in the solute, or do molecules interacting directly with the solute play the most decisive role?
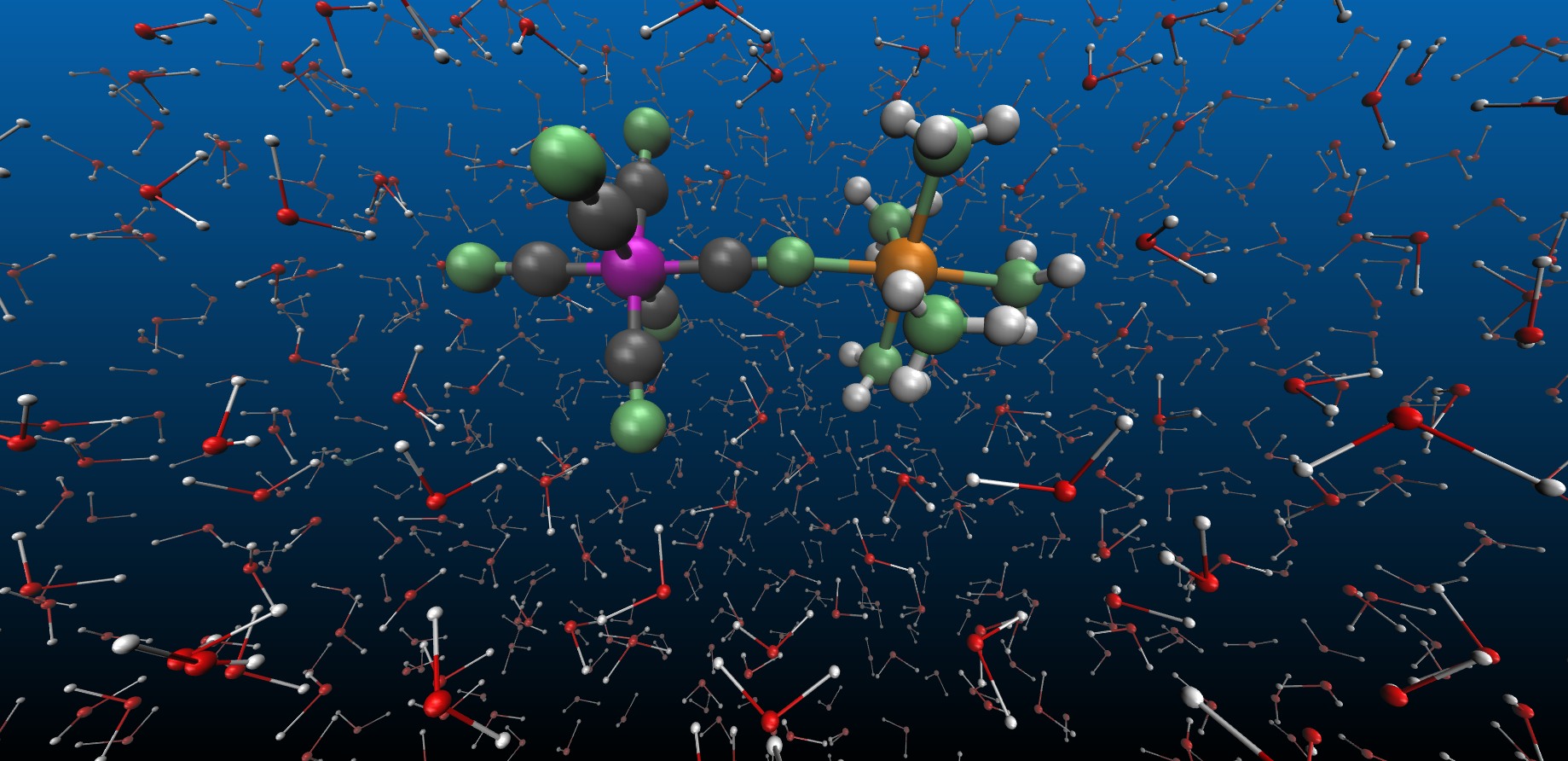
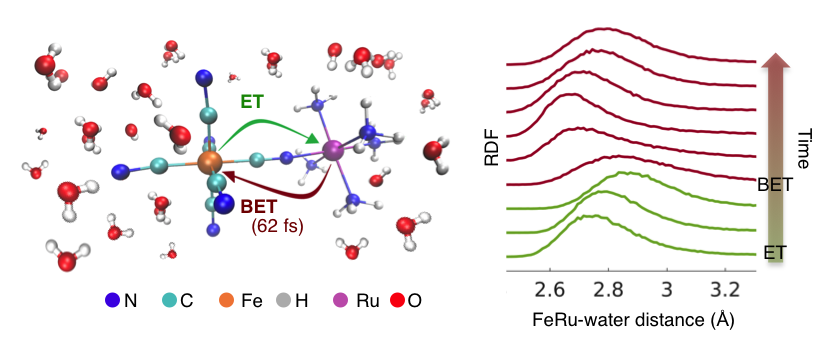
In our work, we have taken a further step in trying to answer these fundamental questions. We have investigated electron transfer between an Fe and Ru metal sites bridged with a cyanide ligand in a mixed valence complex, [NCFeII(CN)5(NH3)5RuIII]-, referred to as FeRu for brevity (Figure 1). The FeRu complex is dissolved in water, where strong hydrogen bonds form between the ligands of FeRu and the water molecules. We utilized femtosecond X-ray pulses available at the Linac Coherent Light Source to probe the structural dynamics coupled to the photoinduced metal-to-metal charge transfer and the subsequent ultrafast (~62 fs) back electron transfer. Elastic X-ray scattering signals were recorded at different time-delays after the optical excitation, allowing us to make a molecular movie of the electron transfer reactions with femtosecond and sub-ångström resolution.
We initially expected that X-ray scattering signals would report on intramolecular changes, as well as on both short-range and long-range solvent reorganization. This is because previous studies have shown that intramolecular vibrations, such as stretching of the cyanide bridge, are coupled to the electron transfer in FeRu [1]. On the other end, the electron transfer between the Fe and Ru atoms changes both the dipole moment of the molecule and the strength of solute-solvent hydrogen bond interactions, and the water molecules would have to reorganize significantly in response to both these changes.
With the help of molecular dynamics simulations, we discovered that, in the short lifetime of the electron transfer process (~62 fs), the solvent molecules in close proximity to the FeRu molecule undergo the largest nuclear displacements and that the X-ray scattering signals reported exclusively on short-range solvation dynamics. Specifically, we monitored the concerted motion of the water molecules hydrogen bonded to FeRu ligands: these molecules translate away from the solute when hydrogen bonding interactions are weakened upon metal-to-metal charge transfer, and then oscillate back to the original position upon back electron transfer (Figure 1).
This is the first time that water motions arising from changes in specific solute-solvent interactions have been directly observed. We achieved these results by combining the structural sensitivity of X-ray solution scattering and the femtosecond resolution offered by X-ray Free Electron Lasers (XFELs). XFELs are currently undergoing high-repetition and high-energy upgrades, which will yield improved structural sensitivity for future X-ray solution scattering experiments. This holds the promise to reveal with unprecedented details the solvent motions driving electron transfer reactions in liquid environments. Our work paves the way to these future studies, and it’s the first step towards a molecular level understanding of solvent-controlled electron transfer reactions. More details of this work can be found at https://www.nature.com/articles/s41557-020-00629-3.
References
[1] Courtney, T. L., Fox, Z. W., Estergreen, L. & Khalil, M. Measuring coherently coupled intramolecular vibrational and charge-transfer dynamics with two-dimensional vibrational-electronic spectroscopy. The Journal of Physical Chemistry Letters 6, 1286–1292 (2015)
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in