Spatial reconstruction of single-cell gene expression data.
Published in Bioengineering & Biotechnology
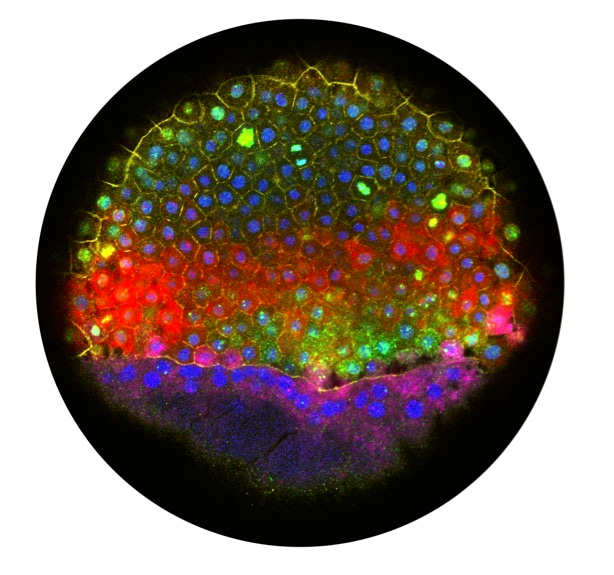

As part of their 25th anniversary collection, Nature Biotechnology invited us to tell the personal story behind our manuscript: Spatial reconstruction of single-cell gene expression data.
How the project first started
Jeff: I was introduced to Aviv Regev and Rahul shortly after I joined Alex Schier’s lab at Harvard; the two labs had already collaborated on previous projects, and Alex suggested that it might be interesting for me to see what we could do together. “You know, now it’s possible to RNA-seq individual cells. What do you think we might learn from that in the embryo?” I was somewhat resistant at first, because I had trained very much as a reductionist during my PhD, but after going over to the Broad to hear some seminars, I was hooked. It seemed like an incredibly powerful approach that was still in its infancy, and not much of the discussion around the Broad was focused on its application to developmental biology. Historically, every time researchers had used a new way to observe development, they had learned new things, and it was clear that this was such an opportunity. Plus, as I got to know Rahul, it quickly became obvious how much fun it was going to be to work together.
Rahul: As a postdoc in Aviv’s lab, I had been working with single-cell RNA-seq for a couple of years, but mostly with in vitro systems. I was excited about the possibility of generating in vivo data, especially as I had spent part of my PhD dissertation working on Drosophila embryogenesis. It was clear that applying single-cell sequencing to understand development and gastrulation was a great opportunity. Also, Schier lab members had a well-deserved reputation of being incredibly fun people who were great to work with. They hosted great lab parties, and I had been looking for opportunities to collaborate with them, with an additional hidden motive of hoping to present at (or at least, be invited to) the Schier lab annual ski retreat.
Focusing on spatial location
Jeff: Spatial information is a crucial determinant of cell fate during development. Commonly in development, cells are instructed by extracellular signals that are produced in different locations. Based on where a cell is, it sees different signals and thereby generates different downstream transcriptional responses. In zebrafish embryos, for example, localized BMP, Nodal, FGF, and Wnt signals determine gene expression patterns and specify the germ layers in the first few hours of development. It was clear that if we could probe the relationship between cells’ spatial position and their gene expression, it was going to be hugely informative.
The first time that Rahul and I met, we spent quite a while talking about what kinds of questions would be interesting to study, and what we might learn from profiling zebrafish embryos. I mentioned this strong dependence on spatial information and showed him a bunch of images of gene expression from the early embryo. I thought it would be amazing to work on more deeply profiling the spatial dependence on gene expression in the embryo. He was immediately on board, because he had already been thinking about ways to combine diverse sources of information for scRNA-seq data (which is now a major focus of his lab) and was excited to try this for spatial information.
Rahul: I do remember that Jeff had produced some astonishing images profiling the early stages of zebrafish gastrulation. Jeff’s excitement for studying this system was contagious as well, and he easily convinced me that to understand fate decisions in early development, we needed to resolve spatial gene expression patterns. At the time, methods for generating transcriptome-wide spatial profiles were in early stages, so mapping single cell RNA-seq profiles seemed like it might be an interesting approach.

Data collection and initial results
Rahul: Given the complexity of cell types in early development, we thought that we’d likely need to profile close to 1,000 cells to get a reasonable spatial map. This was still a major undertaking in 2014 – both for costs and experimental work. I had been working, with the assistance of a Regev Lab research associate Dave Gennert, to automate Rickard Sandberg’s SMART-Seq2 protocol in 96-well plates on an Agilent Bravo liquid handler. The first time we really got the setup to work was all the data that Jeff and Dave collected for this project. On one hand, the robotic setup was invaluable for generating high-throughput data. On the other hand, we had some hiccups.
Jeff: We started with just 8 cells that I had taken from defined locations in the embryo as a pilot. I remember having a couple of slides in one lab meeting just comparing gene expression between them to confirm that what we saw made sense. The title was completely understated — “Single-cell RNAseq: data looks alright.” But even those first 8 cells revealed some localized gene expression that we had not known before. And so, it was off to the races. Aviv’s lab essentially adopted me in a display of incredible generosity – Dave trained me in the protocol he and Rahul had worked out, and I spent many evenings working on spare benches around the lab, making plenty of friends in the process. It was a really exciting and vibrant place to join.
The most experimentally challenging aspect of the project was that we decided to isolate and sequence cells from defined locations in the embryo. At the developmental stage when I would collect the cells, there are no visible characteristics along the dorsal-ventral axis, so I had to label the embryo to keep track of where the cells came from until I could visualize it later. I would transplant a small clump of fluorescent cells into each embryo, place the tip of my transplantation capillary into the embryo and photograph its location relative to the labeled cells, pipette out a few unlabeled cells from nearby, disaggregate them, and lyse one, then image the embryo again later to see where the fluorescent cells were once the dorsal side became apparent. It was a little like being a one-man band: left hand moving the dish with embryos, right hand holding the capillary needle, foot on a pedal to trigger the microscope to take a picture, and of course, the end of the transplantation suction tube in my mouth (it’s a pretty common way to control transplantations among zebrafish researchers). I still remember Rahul’s face when I described this process: “You’re mouth pipetting?!”
Rahul: Most of the data we generated was from dissociated embryos, where we lost all information about the cell’s spatial location and had to infer it computationally. Jeff could dissociate and pick hundreds of cells quickly, and the sequencing experiments for these plates worked beautifully. Some of the most exciting moments for this project involved looking through the early data on these cells, we were thrilled at how much information was in there.
Jeff had also generated one 96-well plate of cells where we knew exactly where the cells came from (‘reference cells’). As described above, this was incredibly tedious work for him — I think he spent more than a week doing nothing else but just isolating and picking these cells, so that we could sequence them and benchmark our algorithm. By far the worst part of this project was the day I had to tell Jeff that the robotic pipeline failed on that specific plate, and he had to go back and collect more reference cells. Even now I’m still afraid when we process a set of rare or irreplaceable samples, and that experience made me even more grateful for experimental collaborators who go through incredible lengths to collect precious samples.
Computational and experimental collaboration
Jeff: I started the project with very little computational experience, and I was devouring online courses in R programming and statistics in my spare time to catch up. I must have driven Rahul crazy with the number of naïve programming and statistics questions I asked him! Still, he was incredibly patient, and I ended up learning so much from working with him. Sometimes he was practically clairvoyant – I once called to ask him about a differential expression test we were using, and before I had even finished asking the question, he was already answering: it was sensitive to the proportion of cells expressing a gene as well as the expression level within non-zero cells.
I tried to turn my programming exercises into useful code for the project; for me, an early highlight of the project was debuting the 3D embryo in situ plotting functions I had written to Rahul and Aviv – it made our preliminary results feel very real, and I think we really all got fired up in that moment to see how far the project can go. By the time we got reviews back on the paper, I had learned so much from working with Rahul that I was able to do many of the revision analyses myself, including the spatial marker bootstrapping. Honestly, it was a collaboration that forever changed my life and my career, as that time spent laid the foundation for the work I would do in the rest of my post-doc on developmental trajectories.
Rahul: There’s something about the nature of single-cell sequencing datasets that makes you want to analyze it yourself. I think by now, there are probably hundreds if not thousands of people who have taught themselves R or python in order to analyze their own scRNA-seq datasets. Jeff learned to program while we were developing Seurat, and as a result, we always aimed to make it accessible and fun to use. Even in writing the first vignettes and tutorials for spatial mapping, we were thinking about how other people would interact with them and utilize our functions in their analysis. Having such a close collaboration with Jeff really helped me with that perspective, and I think that’s had a substantial impact on our documentation and the accessibility of our tools.
New methods and discoveries
Jeff: Understanding gene expression patterns during zebrafish embryogenesis had been a major effort in the community—the Thisse lab had conducted a monumental in situ hybridization screen that identified thousands of gene expression patterns. Our work extended this — it agreed with the published patterns and identified several thousand more, including a catalog of excellent new markers for several tissues. We also identified an unexpected blastula-specific stress response that incorporates developmental regulators in the data; while the stochastic, scattered expression of several of the involved genes had previously been remarked on, it was not clear until our work that they were a coherent program and actually co-expressed in the same cells. Studying this response is actually an effort that is ongoing in my laboratory. We made a huge effort to make the data as available as possible and also very detailed vignettes to demonstrate how we had analyzed the data and how others could do the same. The work paved the way for generating similar catalogs in organisms that had not had the benefit of decades of community input, accelerating research in non-model organisms.
Rahul: One of the interesting analytical challenges for our spatial mapping strategy involved matching up two very different data types: single-cell RNA-seq profiles and in-situ hybridization measurements for 47 ‘landmark’ genes. They had very different noise profiles - in particular due to the extensive sparsity in scRNA-seq for lowly expressed genes - and different statistical properties as well. We developed a regression-based strategy to improve the robustness of the scRNA-seq ‘landmark’ gene measurements based on the overall correlation structure in the data, and fit Gaussian mixture models to identify correspondences between sequencing / FISH profiles. These steps were essential for obtaining accurate spatial maps in our project, but they also represented early strategies for scRNA-seq imputation (i.e. ‘denoising’) and data integration. Those problems have generated enormous interest - and progress - from the computational biology community.
Co-publication with the Marioni Lab
Rahul: While we were generating data, I was invited to EMBL/EBI to give a guest lecture and met John Marioni for the first time. In fact, the chance to meet John was the reason I accepted the invitation in the first place, and we had a great conversation about recently published papers and statistical methods for scRNA-seq. I remember that just before he had to leave, he opened up a paper he was currently writing and proceeded to show me a method to map single-cell RNA-seq profiles to a spatial annelid brain atlas. While the biological system was different, conceptually it was exactly the same idea that we were pursuing in zebrafish. This led to my first experience with manuscript co-submission.
While I was quite nervous at the onset, we learned an enormous amount as Aviv, Alex, and John guided us through the process. John was very generous – even though his manuscript was at a more advanced stage, he waited for over a month for us to finish ours so that we could submit together. Later — when John’s reviews took longer to come back — we delayed formal publication of our manuscript in order to allow the papers to come out together. Even though our papers were on the same topic, it never felt like we were competing, and the co-published stories generated even more interest from the community. This was really a formative experience for me, and my lab has repeatedly co-submitted manuscripts since then, including a second back-to-back publication with the Marioni lab on batch-correction and integration methods.
Jeff: Actually, the co-submission I think lit a fire under us when it was time to write because we were the ones who were behind and holding up the works. We wrote the entire first draft in one sitting. It was a little bit stressful, but surprisingly fun sitting next to each other passing the draft back and forth. It probably also dramatically improved the writing, since our fields of expertise were totally different, and we kept editing down each other’s jargon. We worked through the night and I think met with Aviv and Alex straight away in the morning to show them the draft. Actually, a highlight of the project was getting a very sleep-deprived beer together for lunch after that meeting.
Outlook and impact
Jeff: This work, along with the back-to-back paper from John Marioni’s lab, and the Tomo-seq approach that was published around the same time were some of the very first work in the field of spatial single-cell genomics. It’s a field that has since grown tremendously. It inspired several computational methods for integrating different modalities of data with scRNAseq data (including, but not limited to spatial data) — a field that Rahul’s lab has contributed to significantly. Additionally, the work contained one of the first examples of scRNAseq data that was isolated from defined spatial locations; methods to do this have become very sophisticated, with a number of approaches that use some variation of a slide containing spatial barcodes now in use across the field. Finally, this was an early example of a cell atlas that described a developing organism, an approach that has now become commonplace in developmental biology, and has dramatically accelerated discovery across numerous animals and made characterizing non-model organisms significantly higher throughput.
Rahul: It’s been incredible to see the rapid development of spatial transcriptomics over the past few years, along with computational methods to integrate these data with single-cell RNA-seq. For both Jeff and myself, this project really helped to define our interests and shape our research programs for our future labs. I think I also learned about how I most enjoy working as a scientist, and the importance of identifying collaborators like Jeff and Alex. In New York, I’ve been very fortunate to have close biological collaborations (in particular with Gord Fishell, Dan Littman, Lionel Christiaen, and Peter Smibert), that have been both productive and enormous fun.
After we submitted the paper, Jeff and I were exhausted, and I was very much looking forward to taking some time off to recover. Instead, a couple days later I got an e-mail from another postdoc, Evan Macosko from Steve McCarroll’s lab, who asked if I might be interested to look at some data from a new technology he was developing together with Oni Basu in Aviv’s and Dave Weitz’s lab called Drop-seq. Their astonishing datasets precluded any possibility of rest or skiing, but were a good lesson on how quickly the single-cell sequencing field could (and continues to) rapidly innovate.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in