Stereoselective synthesis of medium lactams enabled by metal-free hydroalkoxylation/stereospecific [1,3]-rearrangement
Published in Chemistry
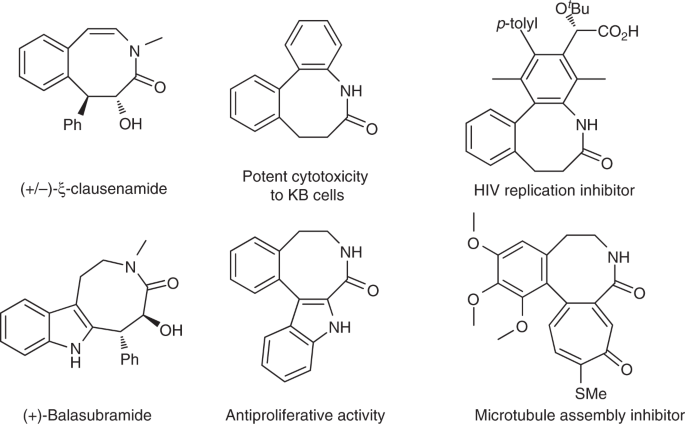
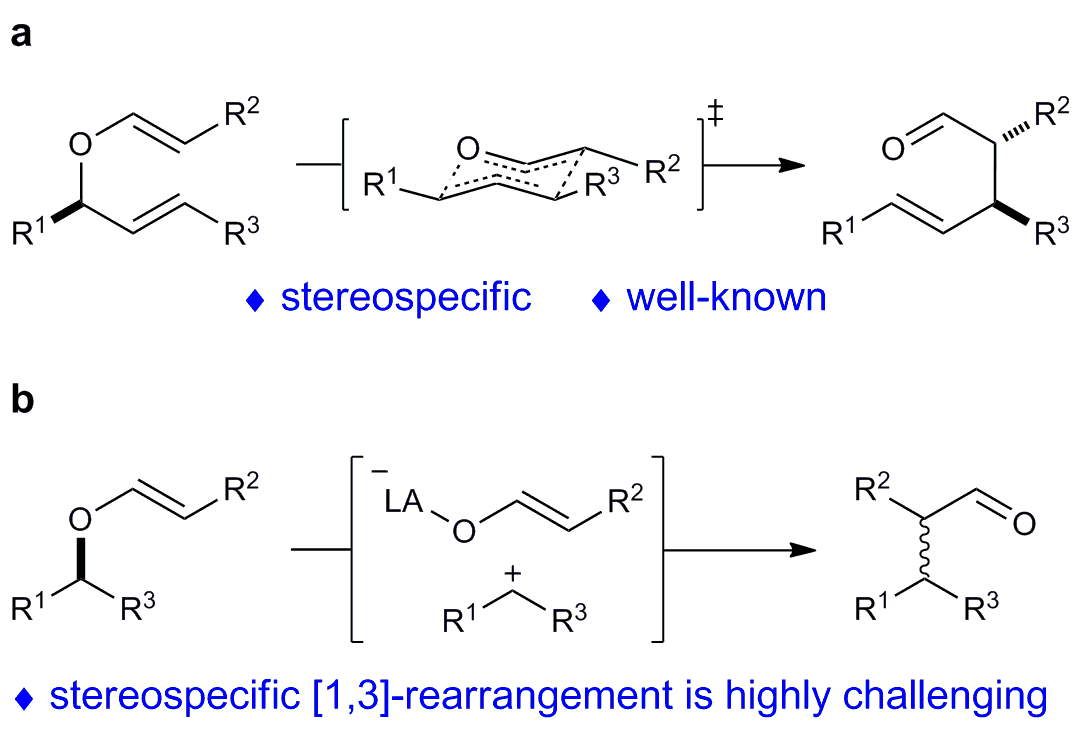
Rearrangement reactions have attracted considerable interest over the past decades due to their high bond-forming efficiency and atom economy in the construction of complex organic architectures. As we know, [3,3]-rearrangement generally proceeds via the chairlike transition state, which makes it stereospecific. Nevertheless, stereospecific [1,3]-rearrangement has been far less investigated because of the uncontrollable presumable zwitterion pairs. The reported examples involving stereochemistry in [1,3]-rearrangement usually lacks generality and significant racemization is observed.
Our group is interested in the development of ynamide chemistry for heterocycle synthesis. Based on our recent study on the yttrium-catalyzed tandem intramolecular hydroalkoxylation/[3,3]-rearrangement, for the synthesis of various valuable medium and large ring lactams (Angew. Chem. Int. Ed. 2017, 56, 4015-4019), we wondered whether we could realize the relevant intramolecular hydroalkoxylation/ [1,3]-rearrangement reaction. Thus, we could construct another kind of eight-membered lactams (benzo[d]azocinones), which are important structural motifs found in many bioactive molecules and natural products. Moreover, we anticipated to control the diastereoselectivity and enantioselectivity of this tandem reaction.
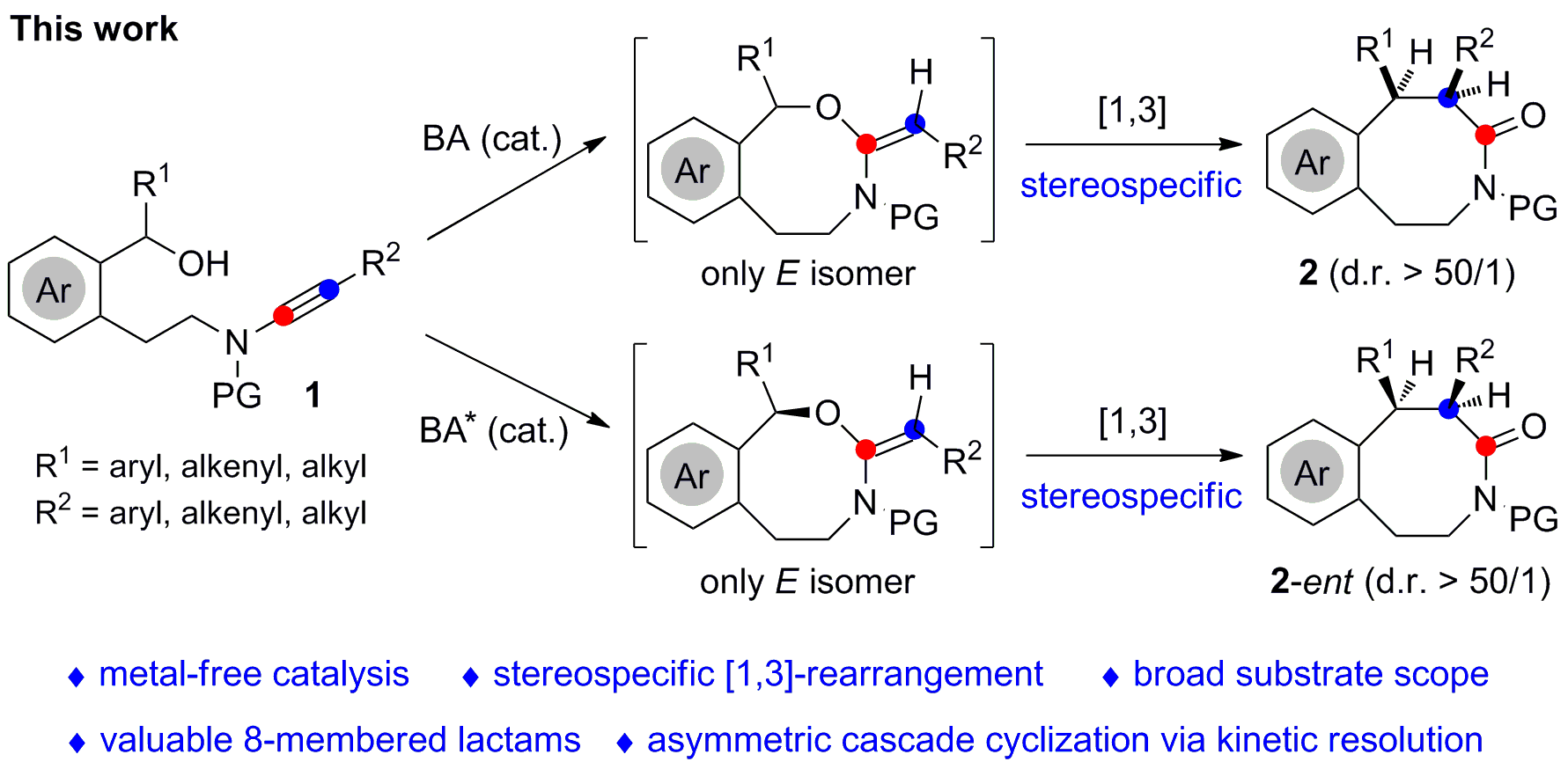
We first tested this hydroalkoxylation/[1,3]-rearrangement reaction with typical gold catalysts such as IPrAuNTf2 and Ph3PAuNTf2, and found that the expected benzo[d]azocinone was indeed formed albeit in low yields. To our delight, the reaction could proceed smoothly with various typical Lewis acid catalysts such as Y(OTf)3, Yb(OTf)3 and Zn(OTf)2. Then, we turned our intention to Brønsted acid catalysts. Interestingly, the desired product was formed in 96% yield with over 50/1 diastereoselectivity in the presence of 0.5 mol % of HOTf as catalyst. Subsequent scope study revealed that a series of benzo[d]azocinones, heterocycle-linked 8-membered ring lactams and 9-membered lactam could be obtained in generally good to excellent yields with excellent diastereoselectivities.
Furthermore, we found that such an asymmetric hydroalkoxylation/ [1,3]-rearrangement was achieved by employing chiral spiro phosphoramide catalyst through parallel kinetic resolution. Preliminary substrate scope study on this asymmetric sequence showed good functional group tolerance. The gram-scale reaction, synthetic transformations and biological tests suggested this cascade cyclization to be potentially useful in synthetic organic chemistry and medicinal chemistry.
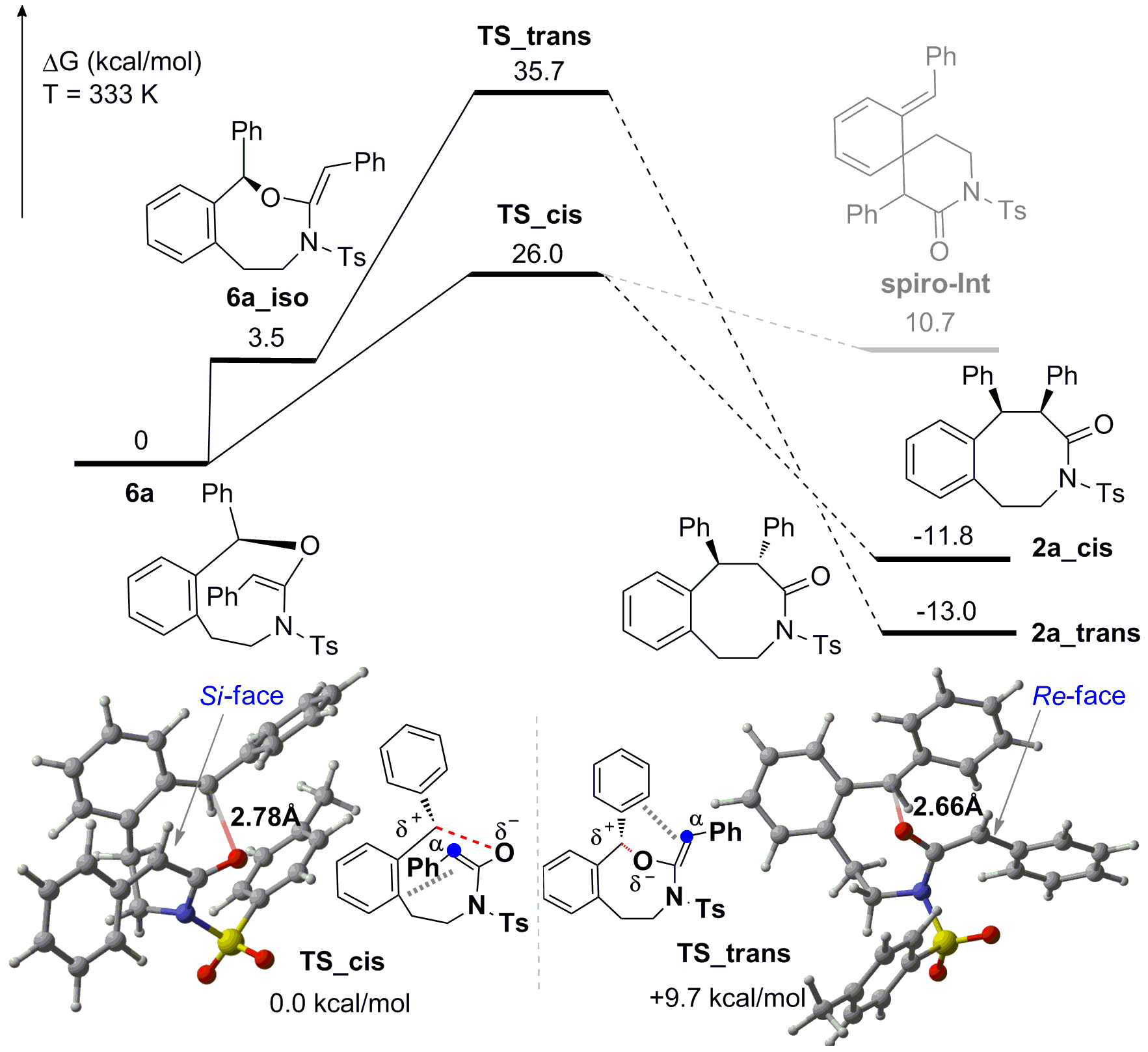
Control experiments and DFT calculations were conducted to further explore the reaction mechanism. The results indicate that the stereospecific and rate-determining [1,3]-rearrangement step is kinetically irreversible and thermodynamically exothermic for the selective synthesis of cis-benzo[d]azocinones. When chiral Brønsted acid is employed, the enantioselectivity is controlled through both ion pairing and H-bonding interactions of keteniminium intermediate with chiral Brønsted acid catalyst.
We hope this novel stereospecific [1,3]-rearrangement involved cascade reaction of ynamides could promote a new wave of research into asymmetric reactions based on rearrangement reactions, chiral Brønsted acid catalysis, and ynamide chemistry, as well as new synthetic route to medium-sized compounds.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in