
Mixed tin-lead halide perovskites are an interesting and promising branch of the traditional lead halide perovskites. These semiconductors exhibit a slightly wider bandgap (~1.2 eV) than silicon, and polycrystalline thin films can readily be fabricated by solution processing techniques. It has been recognized by the perovskite community that during solution processing, tin-based perovskites tend to crystallize faster than Pb-based perovskites, posing a challenge to film quality and device performance. Yet the underlying chemistry deriving this asynchronous behavior between Sn and Pb remains elusive. This puzzle triggered our conception of this work.
A popular view is that the rapid crystallization of Sn perovskites is caused by “the higher Lewis acidity of Sn(II) versus Pb(II)”, however, we found this explanation less than satisfying. Quantitative evaluation of the Lewis acidity can be challenging, and the Lewis acidity of a metal ion has a dependence on the ligands or counterions. As described by the Hard and Soft Acid and Base (HSAB) theory, to react with the soft iodides, Pb(II) seems more thermodynamically favorable than Sn(II) which is considered a hard cation. This subtle difference was confirmed by high-accuracy DFT calculations (pwpb95-d4/def2-tzvpp).
We started to notice the role of lone pair when reading a review article by Aron Walsh and his coworkers (https://pubs.rsc.org/en/content/articlelanding/2011/cs/c1cs15098g), which underlines the interaction between the cation s states and the anion/ligand p states. When binding with DMSO (HOMO -6.4 eV, obtained by b3pw91/def2-tzvpp), DMF (HOMO -6.9 eV), and iodide (5p ~ -7.5 eV), the Pb 6s states (~ -13.5 eV) are too low in energy and thus are relatively inert. While for the higher Sn 5s states (~ -11.5 eV), the Sn 5s-ligand HOMO interaction is strong and the sterically active lone pair can be maintained in the Sn(II)•nL complex.
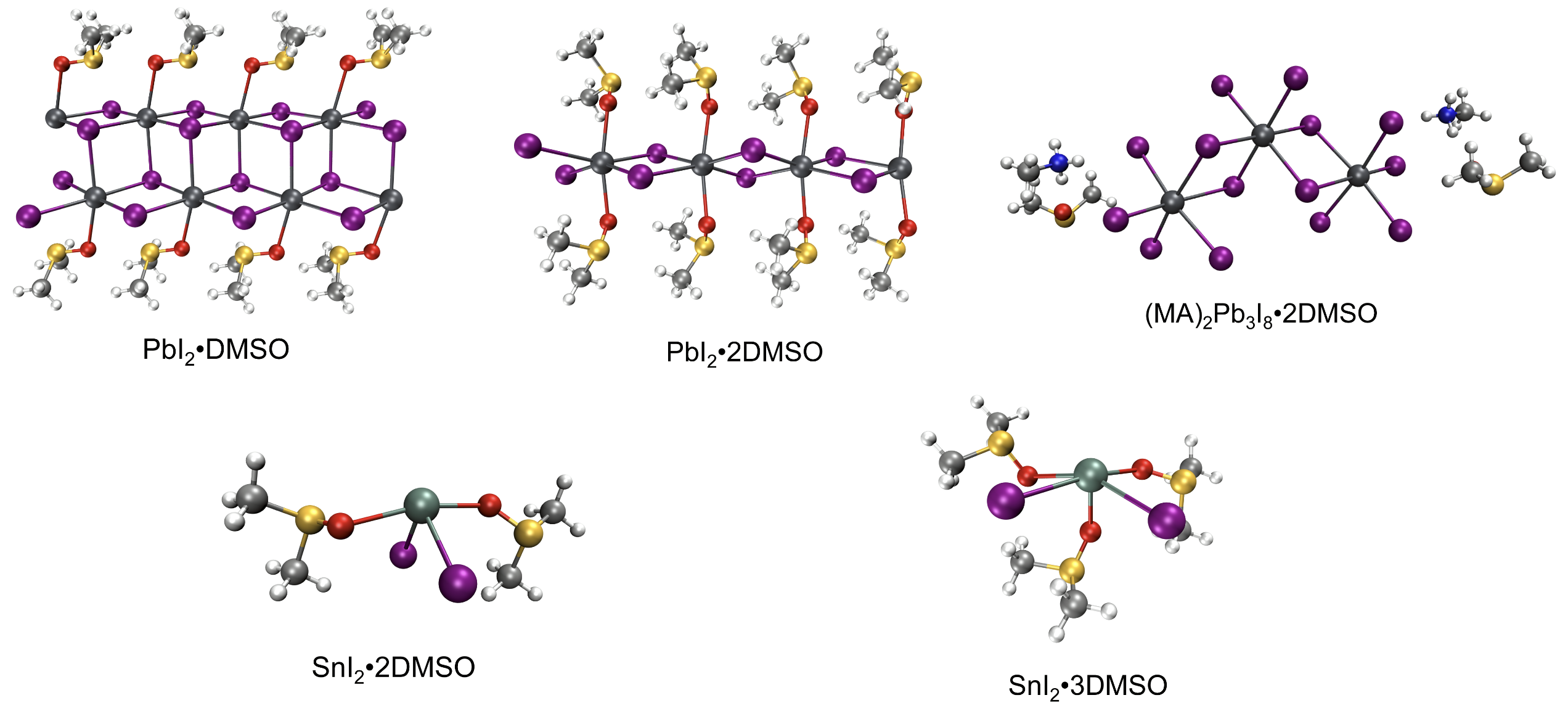
Solvate intermediates of Pb(II) and Sn(II) in perovskite precursor.
We further noticed the structural disparity between the solvate intermediates of PbI2 and SnI2 reported in the literature. For PbI2, the solvates in perovskite precursor tend to form polymeric structures with Pb-I-Pb bridge bonds and hexa-coordination [PbX6] octahedra, like PbI2•DMSO, PbI2•2DMSO, and (MA)2Pb3I8•2DMSO (https://pubs.acs.org/doi/10.1021/jacs.6b04924). While for SnI2, Feng Hao et al. reported an intermediate compound SnI2•3DMSO, which is a molecular crystal packed by isolated SnI2∙3DMSO complex (https://pubs.acs.org/doi/10.1021/jacs.5b06658). More molecular crystals, like SnI2•2DMSO, are determined by Masashi Ozaki et al. (https://pubs.acs.org/doi/10.1021/acsomega.7b01292)
All the above solvate structures were determined by the single-crystal XRD technique, and one could be bothered whether these solid-state structures are maintained in the liquid phase. Extended X-ray absorption fine structure experiments were conducted to detect the local coordination environment of metal cations in the perovskite precursor. The results revealed that there are more iodides in the Pb(II) surrounding and more solvent ligands in the Sn(II) surrounding, consistent with the single-crystal XRD structures reported. One can see how the lone pair stereochemistry impacts the solvate structure of PbI2 and SnI2 in the same perovskite precursor, despite the great chemical similarity shared between Pb(II) and Sn(II). Compared to the polymeric PbI2 solvates, the isolated Sn(II)•nL complexes should be more unstable and prone to decomplexation. In this way, the rapid crystallization of Sn perovskites can be understood.
As for the solar cell fabrication, our group has a fair long history studying lead-based perovskite solar cells in the inverted (p-i-n) style, and we started to do tin-lead perovskites about two years ago. We would like to thank the pioneers in the perovskite community for sharing their invaluable expertise on device fabrication protocols. From the literature, we learned not only great insights into perovskite chemistry but also experimental crafts from labs all over the world. In return, we hope this work can help those devoted to developing perovskite photovoltaics and together we may contribute to a clean environment and future.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in